Our research
The long-term goals of our research are to understand how protein conformational ensembles are reshaped by chemical, genetic, and physical perturbations. We seek to quantify how these perturbations impact protein function and organismal fitness. We are best known for creating multitemperature X-ray data collection approaches, which are especially powerful when paired with multiconformer computational modeling to reveal otherwise inaccessible features of conformational ensembles. Our group integrates high resolution structural biology (X-ray, EM, NMR), functional studies (deep mutational scanning, enzymology), and computation to study biological mechanisms and to improve protein engineering and small molecule discovery. Group members move fluidly between computation and experiment, often inventing new methods to answer their questions.
We work on many different systems and proteins as we find ourselves drawn, again and again, to the tension of high resolution data in structural biology: as resolution gets better, modeling becomes easier… until the resolution gets too good… and then the ensemble begins to reveal itself and the fun challenges begin.
Identifying hidden alternative conformations of proteins and ligands in biophysical data
We study proteins as conformational ensembles. Although X-ray crystallography is an ensemble experiment, the results are typically summarized with a single static structure. We develop software to discover the structural ensembles present in the crystal (or on the EM grid). The ensemble nature of proteins highlighted by this work feeds into all of our mechanistic studies that interpret the functional effects of mutations, that characterize designed and artificially-evolved proteins, or that seek to modulate protein function with small molecules. These methods development efforts are central to discovering new allosteric ligands, through high throughput crystallographic fragment screening efforts. We are expanding, this direction to include modeling and validating protein structural data generated by cryo-electron microscopy (using EMRinger and ensemble modeling) and through integrative approaches to discover cryptic ligand binding sites. These methods development efforts are central to discovering new allosteric ligands, through high throughput crystallographic fragment screening efforts.
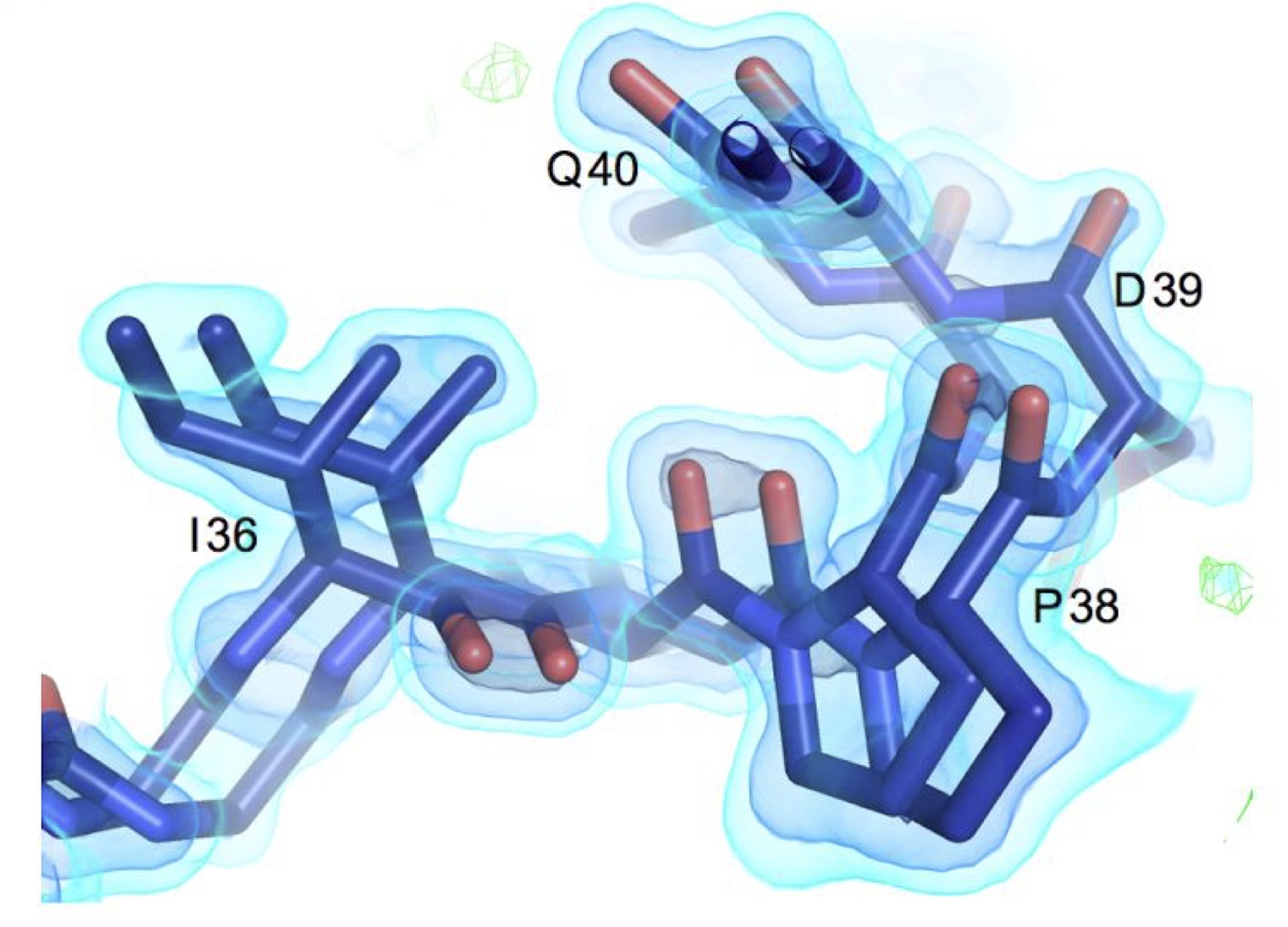
Using multi-temperature X-ray data collection methods in mechanistic studies and ligand discovery campaigns
We recognized that the standard practice of cryocooling crystals could distort protein conformations. In both larger surveys and isolated mechanistic studies, we have demonstrated the value of room temperature data collection for revealing the structural basis of protein conformational dynamics, leading to new insights into enzymes, and increasing connections to dynamics studies from NMR and simulations. Additionally, we have identified how temperature can bias small molecule discovery, leading some fragment sites inaccessible at cryogenic temperatures and repositioning critical water molecules.
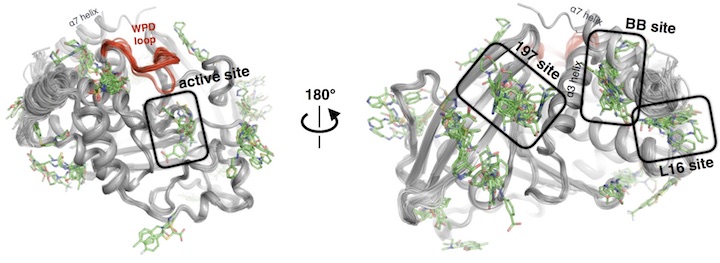
Determining structures that influence microbial-host interactions
We are interested in the structural enzymology and potential therapeutic applications of the human enzymes that degrade chitin, the pervasive polymer that can cause inflammation in the context of allergy and asthma. Using high resolution cryo-electron microscopy (cryo-EM), we are studying the mechanisms of action to newly synthesized antibiotics (in collaboration with the Seiple and Fujimori labs). With this platform, we are also pushing on cryo-EM data processing and refinement methods to enable structure-based drug design, now achieving resolutions better than 2.0 Å. With the improved molecules we generate, we are defining potential resistance mechanisms to these antibiotics using a combination of structural biology and deep mutational scanning experiments. In recent work, we are collaborating with the Bondy-Denomy lab to define the structural basis of anti-CRISPR proteins using a combination of X-ray crystallography, cryo-EM, and NMR. The mechanism of action for many of these proteins remains mysterious.
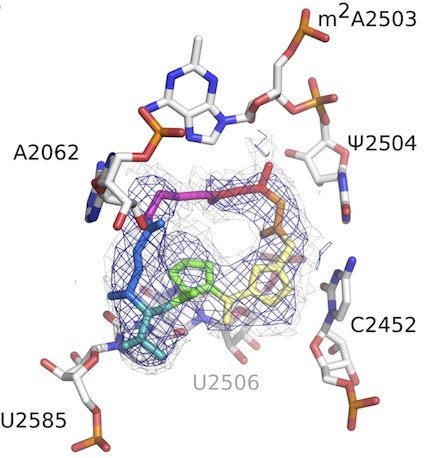
Identifying unifying concepts between systems and structural biology
We are intrigued by the similarities in genetic epistasis and thermodynamic measurements and are applying these insights to large-scale studies of point mutants and posttranslational modifications. We attempt to connect the response to genetic (mutation), chemical (ligands), and physical (temperature) in experiments to define the basis of allosteric regulation and function. We like to bring an evolutionary biology perspective to the table to understand the relative importance of these different perturbations in shaping the proteins we see today.
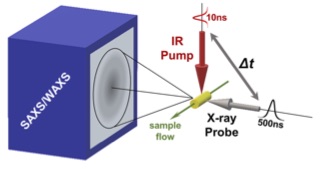
Creating new time-resolved experiments to probe correlated motions in proteins
A major limitation of most biophysical techniques is the inability to directly reveal correlations in motions between distinct regions of macromolecules. We are taking advantage of the new capabilities of next-generation X-ray free electron laser (X-FEL) light sources to perform radiation damage-free imaging of proteins and to watch how protein ensembles respond when perturbed by rapid temperature jumps using the X-FEL. At equilibrium, X-ray diffuse scattering has the potential to reveal these motions; however, we currently lack the ability to collect, integrate, and refine diffuse scattering data. Our long-term goal is to increase the information content of every X-ray diffraction experiment to reveal atomic level coupling at high resolution and improved models of grouped flexibility at low resolution.