Inspecting PanDDA event maps deposited in the Protein Data Bank
Background
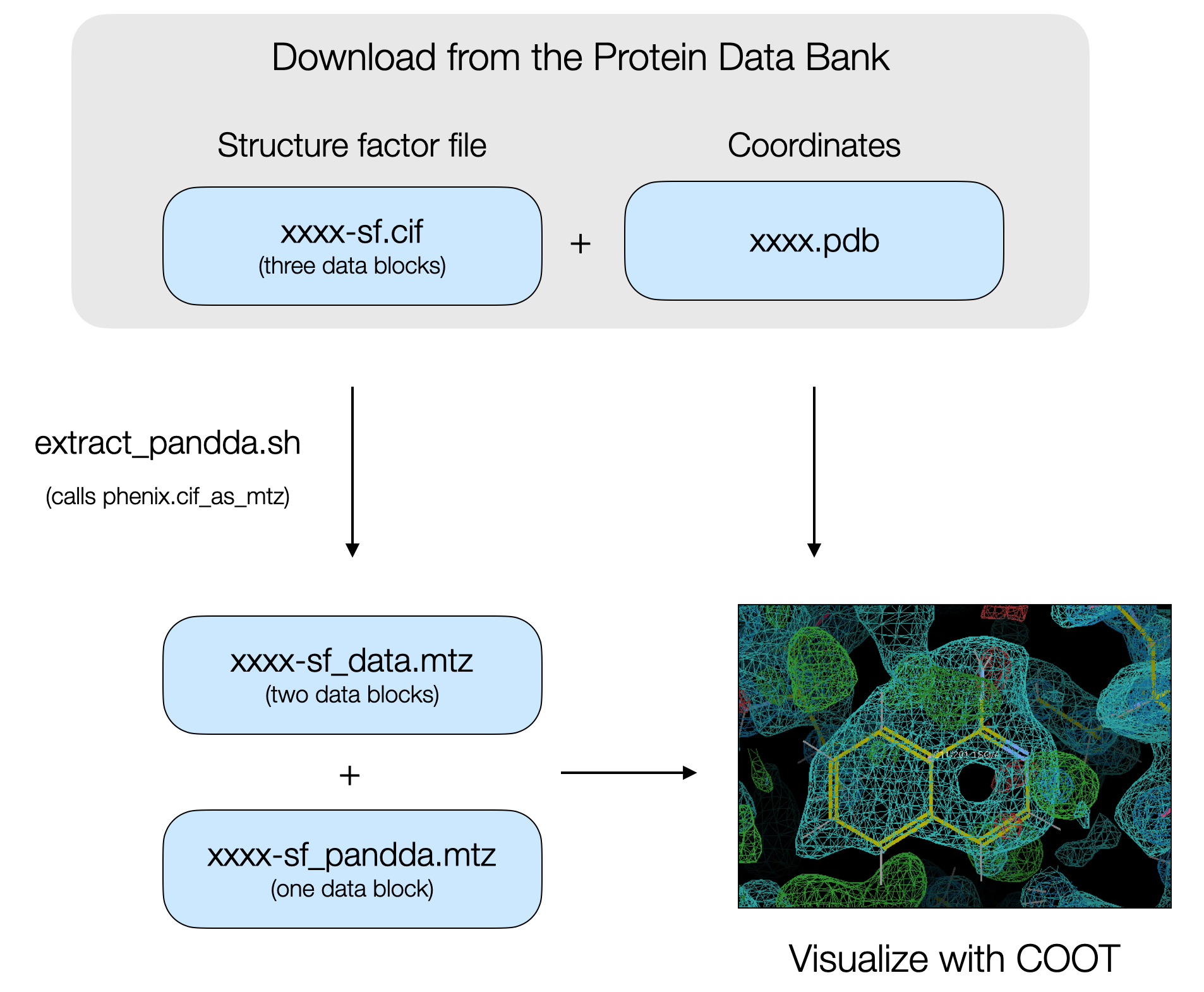
The PanDDA algorithm is a super useful tool for detecting low occupancy ligands in electron density maps obtained by X-ray diffraction. Low occupancy ligands are frequently encountered in fragment screening campaigns, and PanDDA can greatly increase the hit rate of a fragment screen and therefore increase the number of starting points available for fragment-based ligand discovery. We’ve used PanDDA for fragment screens against the PTP1B phosphatase and the NSP3 macrodomain from SARS-CoV-2. After modeling ligands, the data are deposited in the PDB. Data includes the atomic coordinates, the structure factor intensities, the map coefficients after final refinement with the ligand, and the PanDDA event map coefficients. The structure factor intensities and the map coefficients as separate data blocks in a single CIF.
The problems
There are two problems with looking at this data after downloading it from the PDB. The first problem is that because of the low occupancies of ligands, maps based on the structure factor intensities or the map coefficients after final refinement with the partial occupancy ligand often do not contain convincing electron density evidence for the bound ligand. That evidence is best found in the PanDDA map.
The second problem is that CIFs with multiple data blocks can be tricky to convert into MTZ files for visualization in COOT. From my experience, running phenix.cif_as_mtz will lead to the correct conversion of the map coefficients from the refined data, however, the PanDDA event map coefficients may not be converted. The structure factors encoding the PanDDA event map are based on the real space analysis and in space group P1, not the symmetry of the corresponding PDB file.
The solution
We split the CIF containing the three data blocks into separate CIFs. Actually, it’s fine just to extract the PanDDA event map block into one CIF, and move the original and refined data in another. Then run phenix.cif_as_mtz on the separate CIFs, with the correct symmetry flags specified, to convert them into MTZ files.
The extract_pandda.sh script does this for you. Download the coordinates and structure factor file from the Protein Data Bank (xxxx.pdb and xxxx-sf.cif files, where xxxx is the four letter PDB code) and move them to a working directory. On the command line, run ./extract_pandda.sh xxxx-sf.cif SG (where SG is the space group of the crystal). The script will split the CIF into two separate CIFs, containing the refined and original data (xxxx-sf_data.cif) or the PanDDA event map (xxxx-sf_pandda.cif). The script then runs phenix.cif_as_mtz and converts the CIFs to MTZs for visualization in COOT.
Caveats
The script needs the Phenix version dev-4338 to run, available here.
Data blocks need to be named as follows in the CIF (where xxxx is the PDB code):
- Data from final refinement with ligand: data_rxxxxsf
- PanDDA event map: data_rxxxxAsf
- Original data: data_rxxxxBsf