Publications
Filter:
Emergence of binding and catalysis from a designed generalist binding protein
Chen Y*, Bhattacharya S*, Bergmann L*, Correy GJ*, Tan SK, Hou K, Biel JT, Lu L, Bakanas I, Gestwicki JE, Volkov AN, Korendovych IV, Polizzi NF, Fraser JS, DeGrado WF
Nature Chemistry, 2026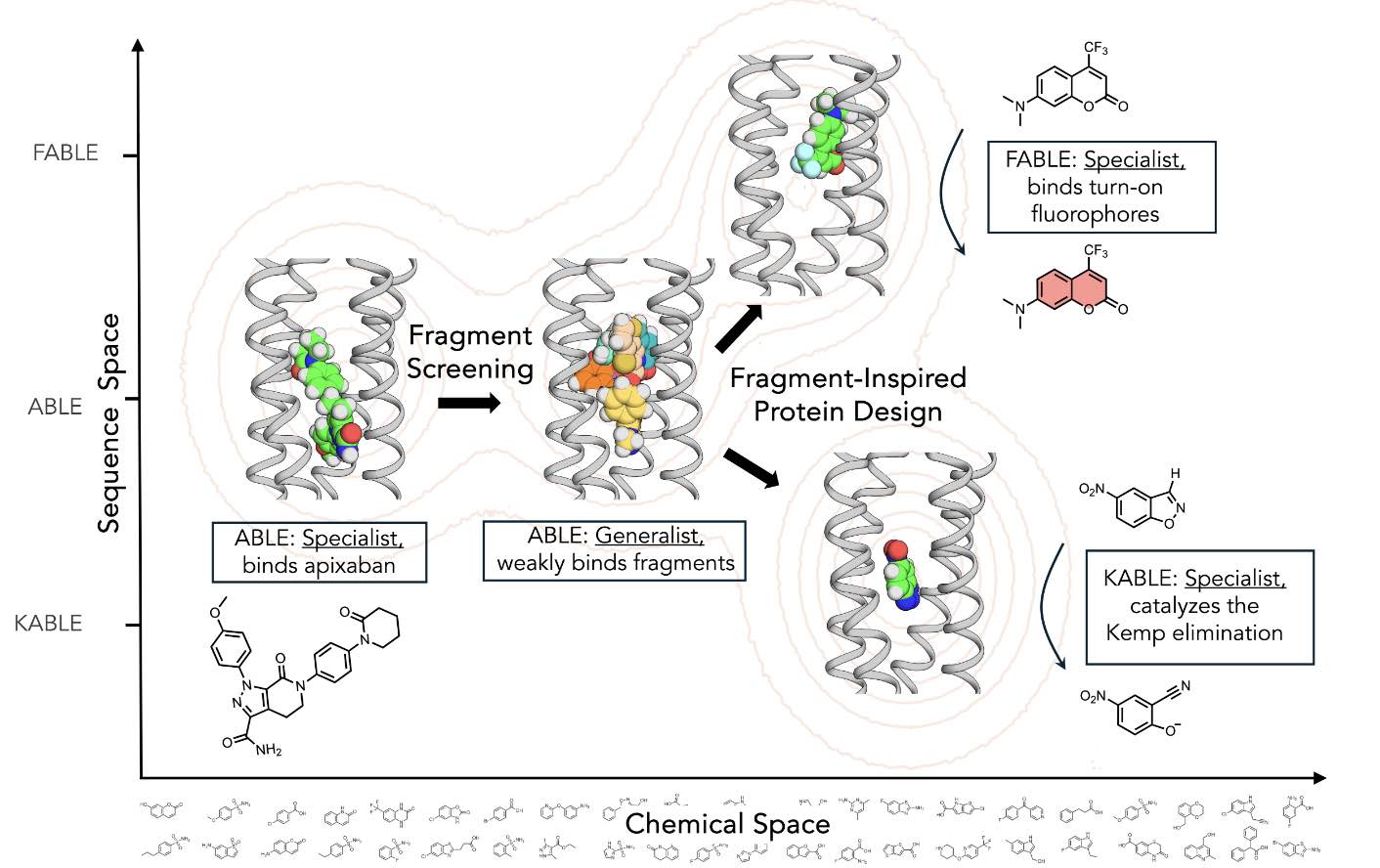
- PMID: 42082787
- BioRxiv Preprint: 635804
- Full Text
- Deposited Structures: 9DW2, 9DWA, 9DWB, 9DWC, 9N0I, 9N0J, 7HIY, 7HIZ, 7HJ0, 7HJ1, 7HJ2, 7HJ3, 7HJ4, 7HJ5, 7HJ6, 7HJ7, 7HJ8, 7HJ9, 7HJA, 7HJB, 7HJC, 7HJD, 7HJE, 7HJF, 7HJG, 7HJH, 7HJI, 7HJJ, 7HJK, 7HJL, 7HJM, 7HJN, 7HJO, 7HJP, 7HJQ, 7HJR, 7HJS, 7HJT, 7HJU, 7HJV, 7HJW, 7HJX, 7HJY, 7HJZ, 7HK0, 7HK1, 7HK2, 7HK3, 7HK4
- Zenodo Record: 13913848 (Structure factor intensities (unmerged, merged, and merged/scaled), PanDDA input and output files including Z-map and event maps in CCP4 format, and refined models including the fragment-bound state extracted from multi-state models)
- SLAC: Researchers pioneer method to rapidly design proteins
- Polizzi Lab
- DeGrado Lab
- Bluetorial Link
Access the paper
Additional Links
Inhibiting the interaction between the mitochondrial receptor Tom70 and SARS CoV 2 Orf9b with small molecules
San Felipe CJ, Verba KA, Krogan NJ, Grabe M, Fraser JS
Biorxiv, 2026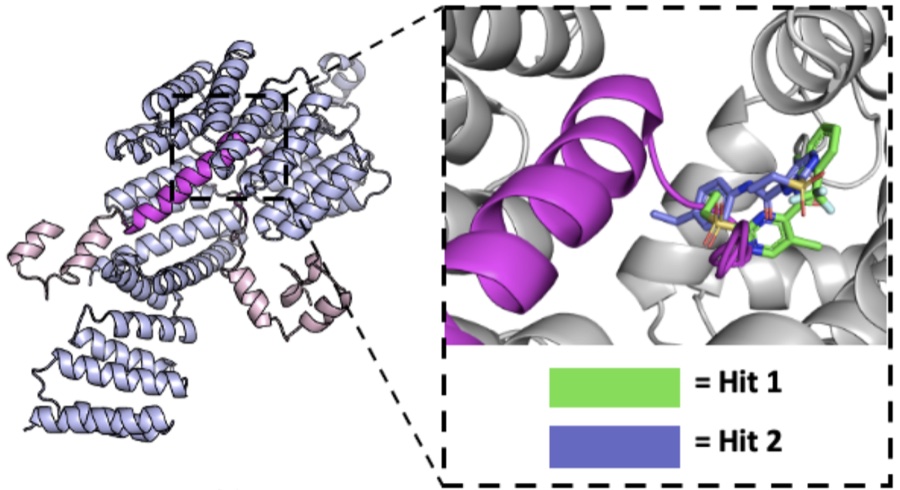
- BioRxiv Preprint: 721040
- Deposited Structures: 13TC, 13TD, 13TE, 13TF, 13TG, 13TH, 13TI, 13TJ, 13TK, 13TL, 13TM, 13TN, 13TO, 13TP, 13TQ, 13TR, 13TS, 13TT, 13TU, 13TV, 13TW, 13TX, 13TY, 13TZ, 13UA, 13UB, 13UC, 13UD, 13UE, 13UF, 13UG, 13UH, 13UI, 13UJ, 13UK, 13UL, 13UM, 13UN, 13UO, 13UP, 13UQ, 13UR, 13US, 13UT, 13UU, 13UV, 13UW, 13UX, 13UY, 13UZ, 13VA
- Zenodo Record: 19750678 (PanDDA analysis of Orf9b fragment screen)
- Grabe Lab @ UCSF
- Krogan Lab @ UCSF
- Verba Lab @ UCSF
Access the paper
Additional Links
Structural adaptations for enhanced translation kinetics in evolved ribosomes
Raskar T*, Costello A*, Badran AH, Fraser JS
Biorxiv, 2026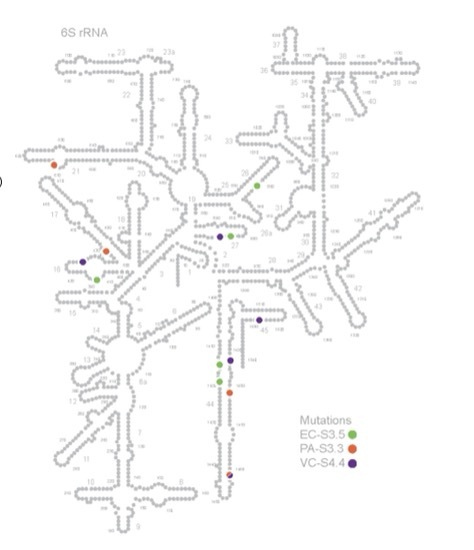
A Computational Community Blind Challenge on Pan-Coronavirus Drug Discovery Data
MacDermott-Opeskin H, Scheen J, Wognum C, Horton JT, West D, Payne AM, Castellanos MA, Colby S, Griffen E, Cousins D, Stacey J, Reid L, Aschenbrenner JC, Fearon D, Balcomb B, Marples P, Tomlinson CWE, Lithgo R, Godoy AS, Winokan M, Barr H, Lahav N, Lavi M, Duberstein S, Cohen G, Fate G, Lefker B, Robinson R, Szommer T, Lynch N, Minh DDL, La VNT, Kang L, Huddleston K, Renslow R, Tollefson M, Walters WP, Xu C, Hsu J, St-Laurent J, Etsmoberg H, Zhu L, Quirke A, Abdul Haleem MI, Alibay I, Baid G, Birnbaum B, Bishop KP, Bohorquez H, Bose A, Brown CJ, Burns J, Cai L, Cedeno R, de Cesco S, Chupakhin V, Clark F, Cole DJ, Corbi-Verge C, Danial M, Davi A, Dehaen W, Doering NP, Dougha A, Dréanic MP, Eakin B, Ehrlich A, Elijosius R, Fülöp J, Gitter A, Goossens K, Gu Y, Head-Gordon T, Hoffer L, Hofmans J, Jiang E, Kaminow B, Khosravi S, Khoualdi AF, Lenselink EB, Liu Z, Liu Y, Liu S, Ma Y, Maher P, Mayer I, Mendez-Lucio O, Mey ASJS, Michel J, Montanari F, Niu T, Ogino R, Palaniappan A, Pan X, Patnaik A, Pham LH, Pinto L, Purnomo J, Rich A, Schaaf L, Schran C, Singh RK, Srilakshmi M, Srivastava SP, Sun K, Sun Z, Talagayev V, Thirukonda Subramanian Balakrishnan B, Titus I, Tkatchenko A, Treyde W, Tricarico G, Tripp A, Vithayapalert N, Wang Y, Wasi AT, Wedig S, Wolber G, Xu B, Zhou W, von Delft F, Lee A, Kirkegaard K, Sjö P, Fraser JS, Chodera JD
Journal of Chemical Information and Modeling, 2026
- PMID: 41749419
- ChemRxiv Preprint: 2025-zd9mr
- Full Text
- Dataset
- Polaris Hub
Access the paper
Additional Links
Structural modification of oxazolidinone antibiotics alters nascent peptide stalling preference and peptide trajectory through the ribosome
Kleinman JI, Raskar T, Klepacki D, Szal T, Vazquez-Laslop N, Mankin AS, Fraser JS, Galonic Fujimori D
Biorxiv, 2026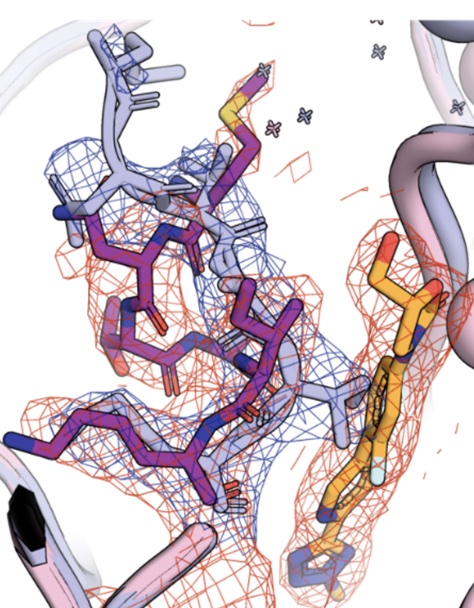
- BioRxiv Preprint: 706219
- Fujimori lab @ UC San Francisco
- Mankin and Vazquez-Laslop lab @ University of Illinois at Chicago
Access the paper
Additional Links
Blind Challenges Let Us See the Path Forward for Predictive Models
Chodera JD, Walters WP, Kosuri S, Fraser JS
Journal of Chemical Information and Modeling, 2026
- PMID: 41651797
- PMCID: PMC12933711
- Preprints.org Preprint: 202512.1130/v1
- Full Text
- Chodera lab @ MSKCC
- Octant
- Pat Walters
- OpenADMET
Access the paper
Additional Links
Structure-based design and synthesis of group A streptogramins that bind to the nascent peptide exit tunnel of the ribosome
Lee IJ*, Li Q*, Raskar T, Pellegrino J, Ecker AK, Howard SY, Fraser JS, Seiple IB
ChemRxiv, 2026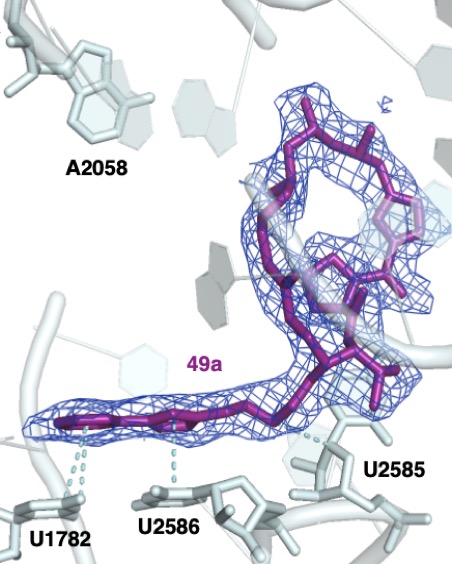
CACHE Challenge #3: Targeting the Nsp3 Macrodomain of SARS-CoV-2
Herasymenko O, Silva M, Correy GJ, Abu-Saleh AAA, Ackloo S, Arrowsmith C, Ashworth A, Ban F, Beck H, Bishop KP, Bohórquez HJ, Bolotokova A, Breznik M, Chau I, Chen Y, Cherkasov A, Dehaen W, Della Corte D, Denzinger K, Doering NP, Edfeldt K, Edwards A, Fayne D, Gentile F, Gibson E, Gokdemir O, Gunnarsson A, Günther J, Irwin JJ, Halborg Jensen J, Harding RJ, Hillisch A, Hoffer L, Hogner A, Hutchinson A, Kandwal S, Karlova A, Koirala K, Kotelnikov S, Kozakov D, Lee J, Lee S, Lessel U, Liu S, Liu X, Loppnau P, Meiler J, Moretti R, Moroz YS, Muvva C, Oprea TI, Paige B, Pandit A, Park K, Poda G, Protopopov MV, Pütter V, Ravichandran R, Rognan D, Rosta E, Sabnis Y, Scott T, Seitova A, Sharma P, Sindt F, Song M, Steinmann C, Stevens R, Talagayev V, Tararina VV, Tarkhanova O, Tingey D, Trant JF, Treleaven D, Tropsha A, Walters P, Wells J, Westermaier Y, Wolber G, Wortmann L, Zheng S, Fraser JS, Schapira M
Journal of Chemical Information and Modeling, 2026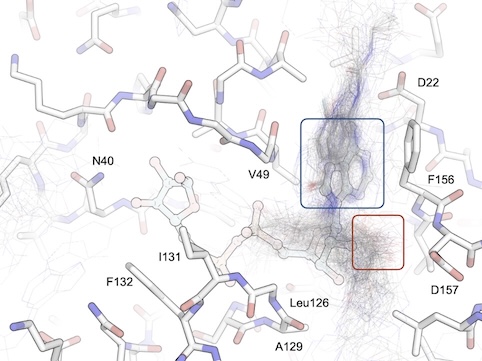
- PMID: 41565251
- PMCID: PMC12892310
- ChemRxiv Preprint: 2025-d1v7v
- Full Text
- Deposited Structures: 7HPI, 7HPJ, 7HPK, 7HPL, 7HPM, 7HPN, 7HPO, 7HPP, 7HPQ, 7HPR, 7HPS, 7HPT, 7HPU, 7HPV, 7HPW, 7HPX, 7HPY, 7HPZ, 7HQ0, 7HQ1, 7HQ2, 7HQ3, 7HQ4, 7HQ5, 7HQ6, 7HQ7, 7HQ8, 7HQ9, 7HQA, 7HQB, 7HQC, 7HQD, 7HQE, 7HQF, 7HQG, 7HQH, 7HQI, 7HQJ, 7HQK, 7HQL, 7HQM, 7HQN, 7HQO, 7HQP
- CACHE Challenge #3
- SGC Toronto
Access the paper
Additional Links
Evaluating beta-tubulin variants as predictors of benzimidazole resistance across Caenorhabditis nematodes
Shaver AO, McKeown R, Reyes Otero JM, Collins JB, Hogan DW, Fraser JS, Dreyer S, Ragsdale EJ, Andersen EC
Biorxiv, 2026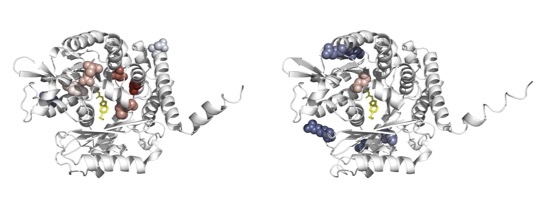
- BioRxiv Preprint: 643047
- Andersen Lab @ Johns Hopkins
Access the paper
Additional Link
Large scale prospective evaluation of co-folding across 557 Mac1-ligand complexes and three virtual screens
Kim J*, Correy GJ*, Hall BW*, Rachman MM, Mailhot O, Togo T, Gonciarz RL, Jaishankar P, Neitz RJ, Hantz ER, Doruk YU, Stevens MGV, Diolaiti ME, Reid R, Gopalkrishnan S, Krogan NJ, Renslo AR, Ashworth A, Shoichet BK, Fraser JS
Biorxiv, 2025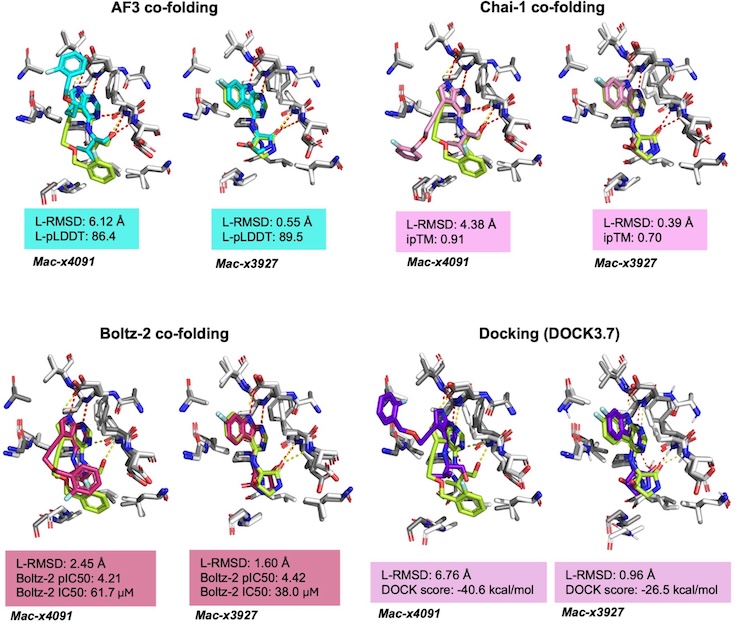
- BioRxiv Preprint: 696505
- GitHub Repository: jongbin99/Cofolding (Scripts and pipelines for the co-folding benchmarks and analysis)
- Shoichet lab
- Know when to co-fold'em
Access the paper
Additional Links
Discovery of AVI-6451, a Potent and Selective Inhibitor of the SARS-CoV-2 ADP-Ribosylhydrolase Mac1 with Oral Efficacy in vivo
Jaishankar P*, Correy GJ*, Matsui Y*, Togo T*, Rachman MM, Stevens MGV, Hantz ER, Zheng J, Diolaiti ME, Montano M, Taha TY, Rosecrans J, Pampel J, Krogan NJ, Shoichet BK, Ashworth A, Ott M, Fraser JS, Renslo AR
Journal of Medicinal Chemistry, 2025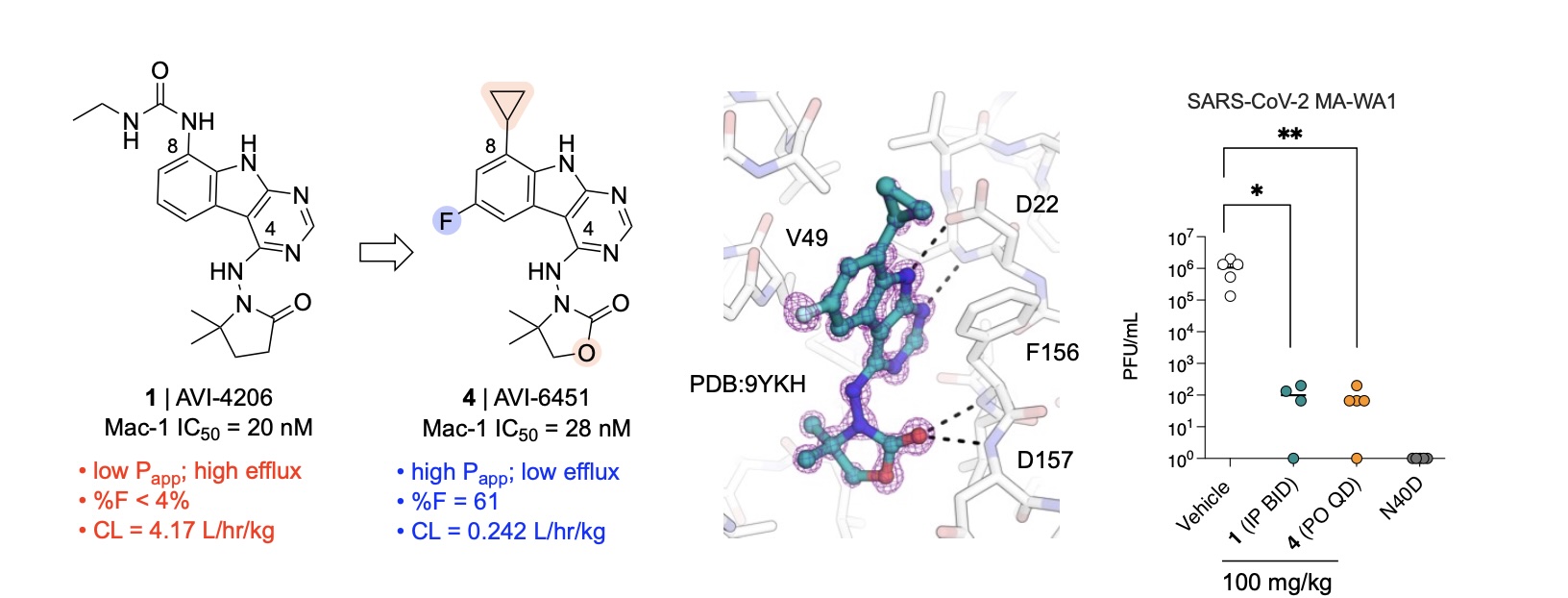
OpenADMET: Embracing the Avoid-Ome to Transform Drug Discovery
Fraser JS, Edgar S, Handly LN, Kosuri S, Chodera JD, Murcko M, Walters WP
Preprints.org, 2025
- Preprints.org Preprint: 202512.1202/v1
- OpenADMET
- Octant
- Chodera lab @ MSKCC
- Pat Walters
Access the paper
Additional Links
Autoregulation of the MET receptor tyrosine kinase by its intracellular juxtamembrane domain
Linossi EM, Espinoza CA, Estevam GO, Fraser JS, Jura N
Biochemical Journal, 2025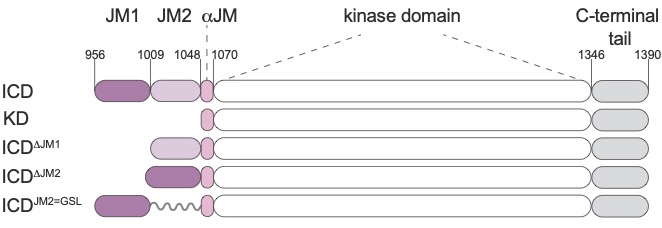
- PMID: 41383124
- PMCID: PMC12751062
- BioRxiv Preprint: 683305
- Full Text
- Jura Lab
Access the paper
Additional Link
Crystallographic Ensembles Reveal the Structural Basis of Binding Entropy in SARS-CoV-2 Macrodomain
Seo L, Farran I, Aslam A, Li X, Jaishankar P, Ashworth A, Fraser JS, Renslo AR, Wankowicz SA
Biorxiv, 2025
- BioRxiv Preprint: 690589
- Deposited Structures: 13VB, 13VC
- GitHub Repositories: Wankowicz-Lab/ensemble_bioinformatic_toolkit (Ensemble bioinformatic toolkit for Mac1 crystallographic analysis), ExcitedStates/qfit-3.0 (qFit 3.0 multiconformer modeling for protein and ligand density)
- Wankowicz lab @ Vanderbilt
- Renslo lab @ UC San Francisco
Access the paper
Additional Links
The Mac1 ADP-ribosylhydrolase is a Therapeutic Target for SARS-CoV-2
Suryawanshi RK*, Jaishankar P*, Correy GJ*, Rachman MM*, O’Leary PC*, Taha TY*, Matsui Y*, Zapatero-Belinchón FJ, McCavitt-Malvido M, Doruk YU, Stevens MGV, Diolaiti ME, Jogalekar MP, Chen H, Richards AL, Kongpracha P, Bali S, Montano M, Rosecrans J, Matthay M, Togo T, Gonciarz RL, Gopalkrishnan S, Neitz RJ, Krogan NJ, Swaney DL, Shoichet BK, Ott M, Renslo AR, Ashworth A, Fraser JS
eLife, 2025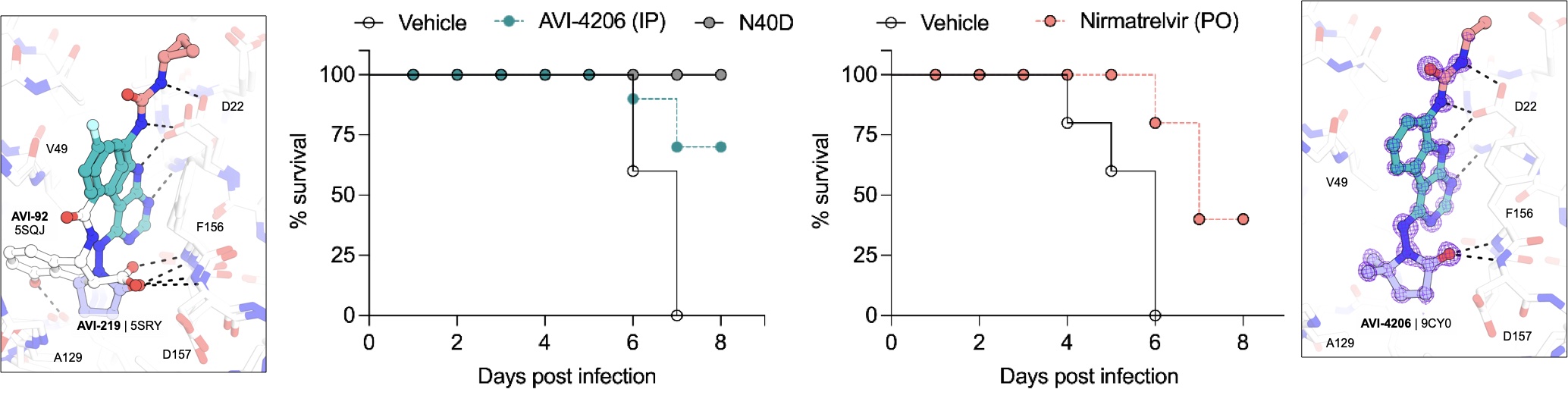
- PMID: 41258893
- PMCID: PMC12629595
- BioRxiv Preprint: 606661
- Full Text
- Deposited Structures: 9CXY, 9CXZ, 9CY0, 7HC4, 7HC5, 7HC6, 7HC7, 7HC8, 7HC9, 7HCA
- QCRG AViDD Program
- Renlso lab @ UCSF
- Ott lab @ Gladstone
- Alan Ashworth @ UCSF
Access the paper
Additional Links
Macrodomain ADP-ribose binding but not ADP-ribosylhydrolase activity is critical for chikungunya virus infection of Aedes mosquitoes
Bardossy ES*, Bergmann L*, Henrion-Lacritick A, Nigg J, Correy GJ, Ashworth A, Fraser JS, Saleh MC
Biorxiv, 2025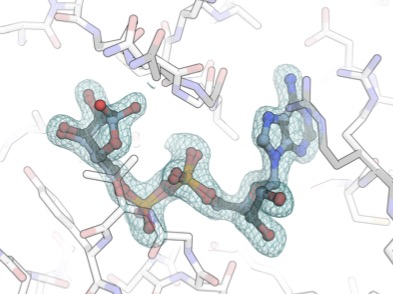
Bayesian multi-state multi-condition modeling of a protein structure based on X-ray crystallography data
Hancock M, Holton JM, Fraser JS, Adams PD, Sali A
Biorxiv, 2025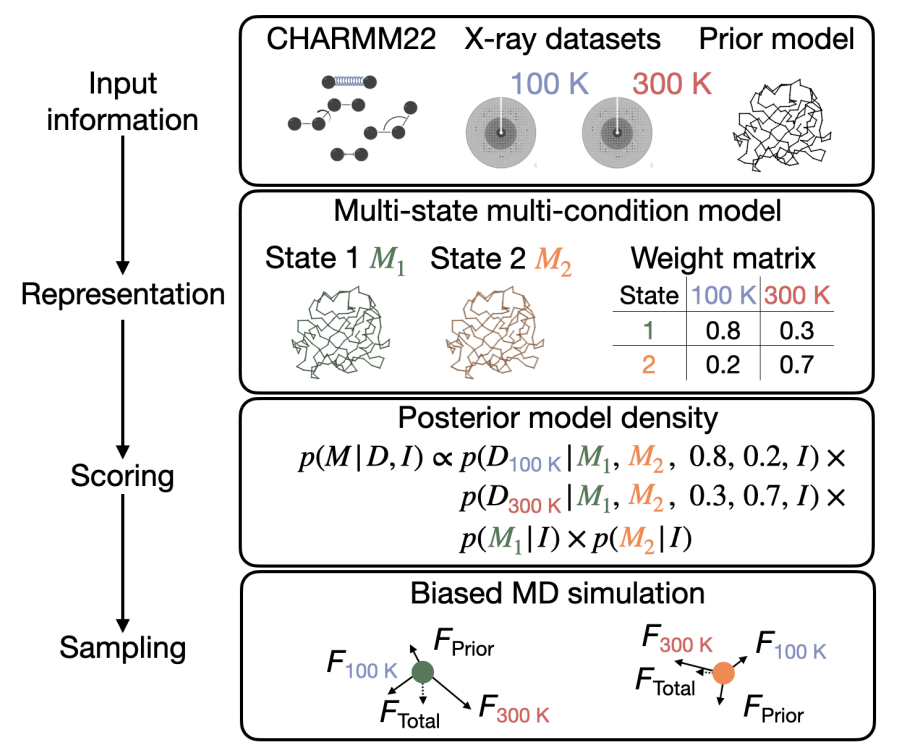
Distal mutations enhance catalysis in designed enzymes by facilitating substrate binding and product release
Zarifi N, Asthana P, Doustmohammadi H, Klaus C, Sanchez J, Hunt SE, Rakotoharisoa RV, Osuna S, Fraser JS, Chica RA
Nature Communications, 2025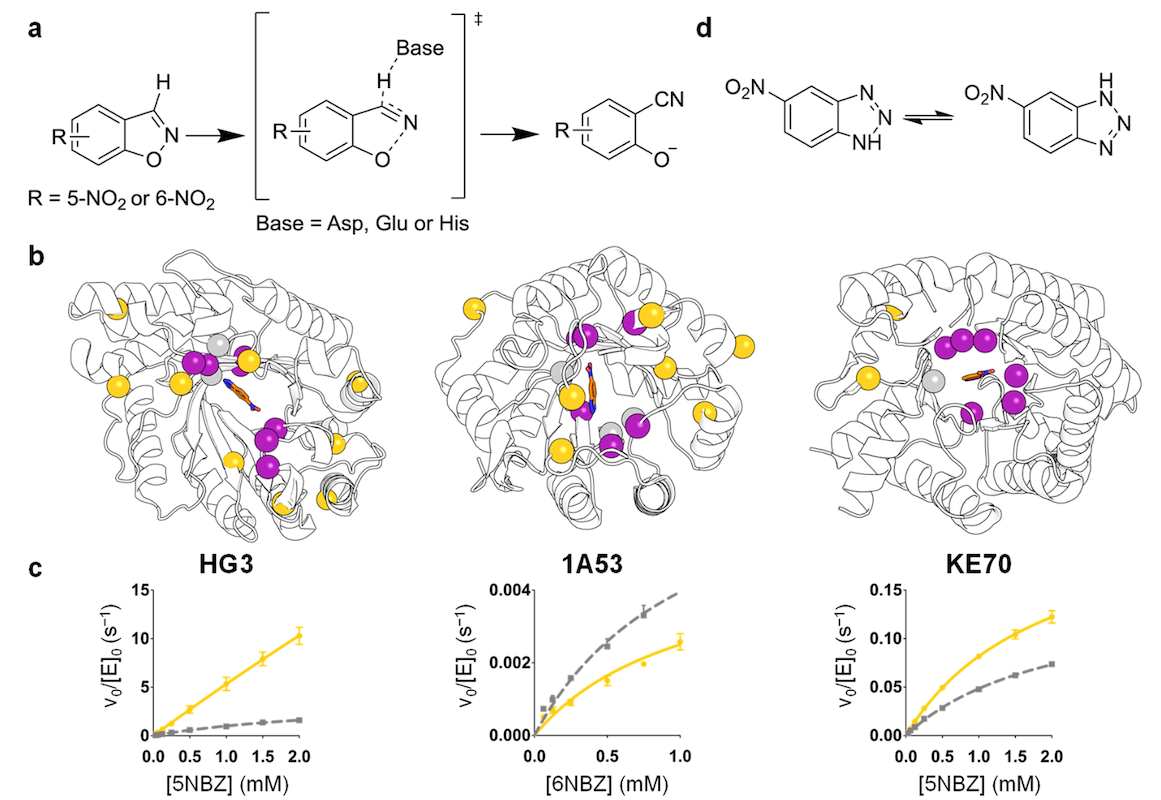
- PMID: 41027962
- PMCID: PMC12484545
- BioRxiv Preprint: 639315
- Full Text
- Deposited Structures: 8FMC, 8FOQ, 8FMD, 8FOR, 8FME, 8FOS
- Chica lab @ University of Ottawa
- Bluetorial Link
Access the paper
Additional Links
Hybrid antibiotics targeting the bacterial ribosome
Yeon SK, Pellegrino J, Raskar T, Tran MLN, Dandan M, Guérin F, Einsiedler M, Cattoir V, Fraser JS, Seiple IB
ACS Central Science, 2025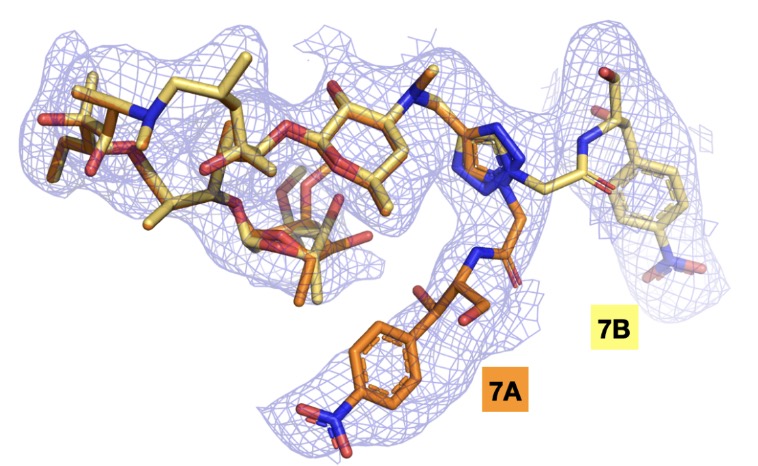
Rosace-AA: Enhancing Interpretation of Deep Mutational Scanning Data with Amino Acid Substitution and Position-Specific Insights
Rao J, Wang M, Howard MK, Macdonald CB, Fraser JS, Coyote-Maestas W, Pimentel H
Bioinformatics Advances, 2025
- PMID: 41550255
- PMCID: PMC12809776
- BioRxiv Preprint: 632281
- Full Text
- GitHub Repository: pimentellab/rosace-aa (Rosace-AA)
- Coyote-Maestas Lab
- Pimentel Lab
Access the paper
Additional Links
Coupled equilibria of dimerization and lipid binding modulate SARS Cov 2 Orf9b interactions and interferon response
San Felipe CJ, Batra J, Muralidharan M, Malpotra S, Anand D, Bauer R, Verba KA, Swaney DL, Krogan NJ, Grabe M, Fraser JS
eLife, 2025
- PMID: 40960494
- PMCID: PMC12443476
- BioRxiv Preprint: 638509
- Full Text
- Deposited Structures: 9MZB, 9N55
- Grabe Lab
- Bluetorial Link
Access the paper
Additional Links
Initial leads to combat streptogramin resistance generated from X-ray fragment screening against VatD
Asthana P, Lee S, MacDonald CM, Seiple IB, Fraser JS
Structure, 2025
- PMID: 40945513
- PMCID: PMC12435910
- BioRxiv Preprint: 635826
- Deposited Structures: 7HZV, 7HZW, 7HZX, 7HZY, 7HZZ, 7I00, 7I01, 7I02, 7I03, 7I04, 7I05, 7I06, 7I07, 7I08, 7I09, 7I0A, 7I0B, 7I0C, 7I0D, 7I0E, 7I0F, 7I0G, 7I0H, 7I0I, 7I0J, 7I0K, 7I0L, 7I0M, 7I0N, 7I0O, 7I0P, 7I0Q, 7I0R, 7I0S, 7I0T, 7I0U, 7I0V, 7I0W, 7I0X, 7I0Y, 7I0Z, 7I10, 7I11, 7I12
- Zenodo Record: 14775497 (Background DMSO datasets of VatD crystals soaked in 10% DMSO for PanDDA analysis used in the study of VatD complexed with fragments.)
- Seiple Lab
- Bluetorial Link
Access the paper
Additional Links
Structure-based discovery of inhibitors of Mac1 domain of nonstructural protein-3 of SARS-CoV-2 by machine learning-augmented screening of chemical space
Ban F*, Ravichandran R*, Correy GJ, Herasymenko O, Silva M, Ackloo S, Bolotokova A, Chau I, Gibson E, Harding R, Hutchinson A, Loppnau P, Fraser JS, Schapira M, Cherkasov A, Gentile F
Biorxiv, 2025
- BioRxiv Preprint: 674529
- Deposited Structure: 7HPW
- CACHE Challenge #3
- SGC Toronto
- Gentile lab @ University of Ottawa
Access the paper
Additional Links
AcrIF11 is a potent CRISPR-specific ADP-ribosyltransferase encoded by phage and plasmid
Chen DF*, Roe LT*, Li Y, Borges AL, Zhang JY, Babbar P, Maji S, Stevens MGV, Correy GJ, Diolaiti ME, Smith DH, Ashworth A, Stroud RM, Kelly MJS, Bondy-Denomy J, Fraser JS
mBio, 2025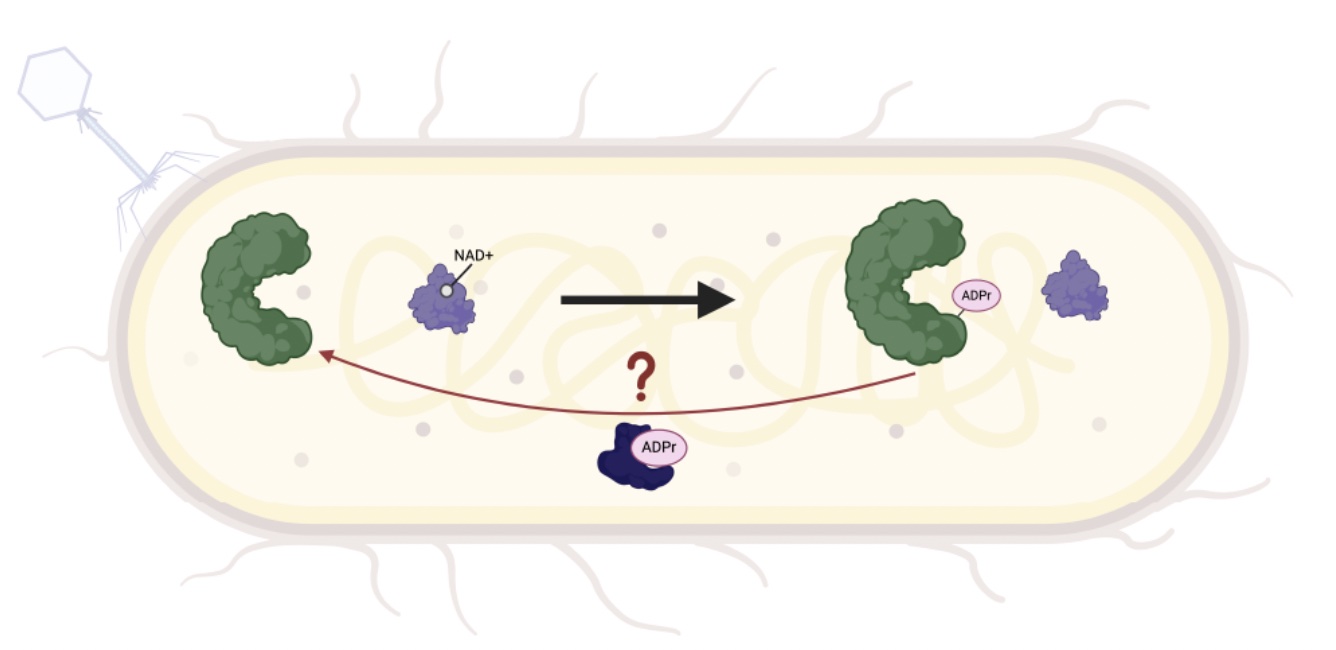
- PMID: 40810510
- PMCID: PMC12421852
- BioRxiv Preprint: 609590
- Full Text
- Deposited Structure: 8DWQ
- Bondy-Denomy lab @ UC San Francisco
Access the paper
Additional Link
Platform for the synthesis and evaluation of verrucarol-based ribosome inhibitors
Tran MLN, Raskar TB, Toth ED, Zhang J, Ecker AK, Yeon SK, Tackie-Yarboi E, DeGrado WF, Fraser JS, Seiple IB
ChemRxiv, 2025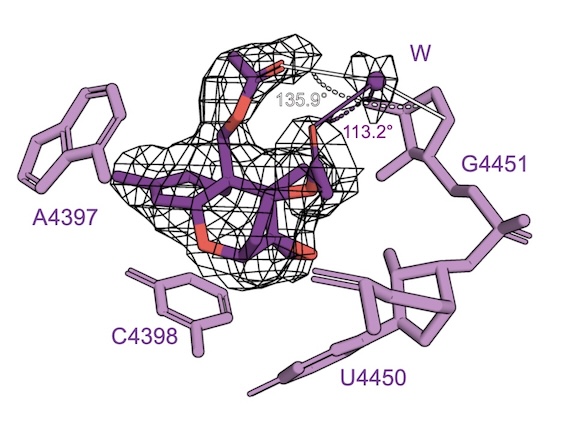
- ChemRxiv Preprint: 2025-7n89p
- Deposited Structure and Map: 9P7T/71353
- Seiple Lab @ Scripps
Access the paper
Additional Link
Coronavirus protein interaction mapping in bat and human cells identifies molecular and genetic switches for immune evasion and replication
Batra J, Rutkowska M, Zhou Y, Ye C, Adavikolanu R, Young JM, Anand D, Verma S, Gordon M, Malpotra S, Cupic A, Kehrer T, Moen JM, Winters DM, Rojc A, Mena I, Aslam S, Martinez-Romero C, Conde Vinas I, Khalil Z, Farrugia K, Banerjee A, Tussia-Cohen D, Dos Santos M, Maji S, Muralidharan M, Foussard H, Chen IP, Fuchs R, San Felipe CJ, Zuliani-Alvarez L, Choudhury P, Obernier K, Gracias S, Suryawanshi R, Ibanez C, Juste J, Pache L, Taha TY, Jouvenet N, Verba KA, Fraser JS, Demeret C, Stroud RM, van Bakel H, Ott M, Hagai T, Polacco B, Swaney DL, Echeverria Riesco I, Bouhaddou M, Eckhardt M, Malik HS, Martinez-Sobrido L, Miorin L, Garcia-Sastre A, Krogan NJ
Biorxiv, 2025
- BioRxiv Preprint: 666918
- Krogan Lab
Access the paper
Additional Link
Product-stabilized filamentation by human glutamine synthetase allosterically tunes metabolic activity
Greene ER, Muniz R, Yamamura H, Bajaj P, Lee DJ, Thompson EM, Arada A, Lee GM, Bonomi M, Kollman JM, Fraser JS
Biorxiv, 2025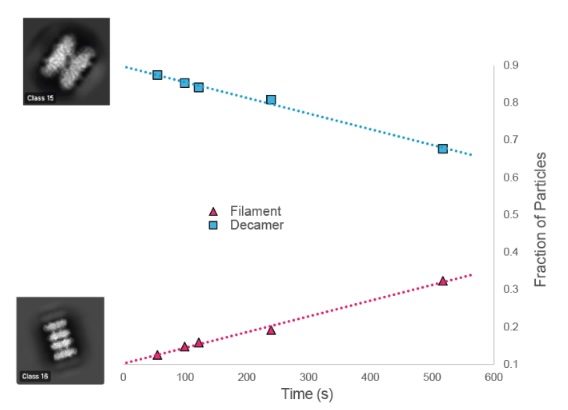
- BioRxiv Preprint: 663231
- Deposited Structures and Maps: 9OTM/70841, 9OTN/70842, 9OTO/70843, 9OTP/70844, 9OTQ/70845
- Zenodo Record: 15742546 (Glutamine synthetase cryo-EM EMMIVox ensemble refinements)
- PLUMED-NEST entry: Product-stabilized filamentation by human glutamine synthetase allosterically tunes metabolic activity
- Kollman Lab @ UW
- Bonomi Lab @ Pasteur Institute
Access the paper
Additional Links
Expanding Automated Multiconformer Ligand Modeling to Macrocycles and Fragments
Flowers J, Echols N, Correy G, Jaishankar P, Togo T, Renslo AR, van den Bedem H, Fraser JS, Wankowicz SA
eLife, 2025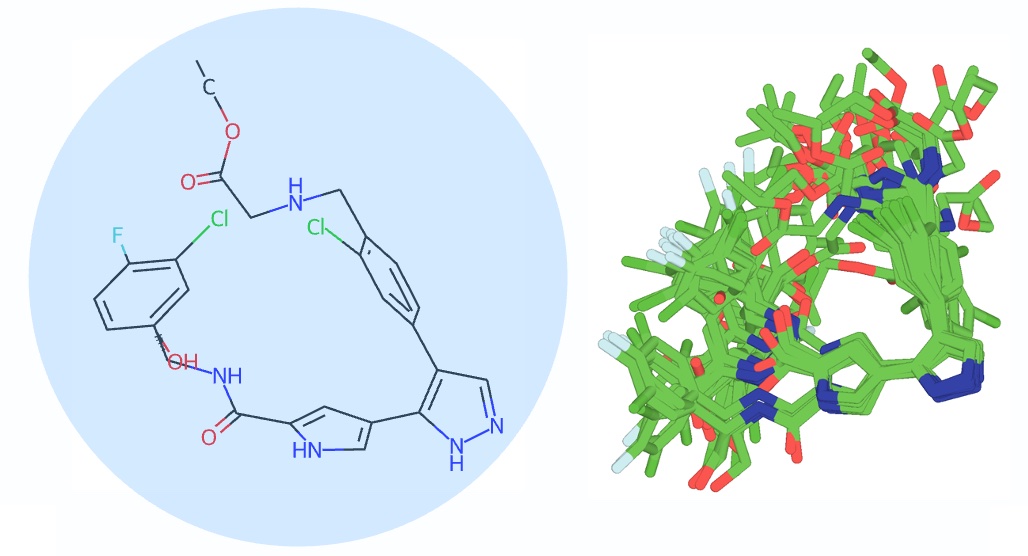
- PMID: 40586518
- PMCID: PMC12208665
- BioRxiv Preprint: 613996
- Full Text
- Deposited Structures: 7HHS, 7HHT, 7HHU, 7HHV, 7HHW, 7HHX, 7HHY, 7HHZ, 7HI0, 7HI1, 7HI2, 7HI3, 7HI4, 7HI5, 7HI6, 7HI7
- GitHub Repository: ExcitedStates/qfit-3.0 (qFit)
- Wankowicz Lab @ Vanderbilt
- Renlso lab @ UCSF
Access the paper
Additional Links
Extensive exploration of structure activity relationships for the SARS-CoV-2 macrodomain from shape-based fragment merging and active learning
Correy GJ*, Rachman MM*, Togo T, Gahbauer S, Doruk YU, Stevens MGV, Jaishankar P, Kelley B, Goldman B, Schmidt M, Kramer T, Ashworth A, Riley P, Shoichet BK, Renslo AR, Walters WP, Fraser JS
Science Advances, 2025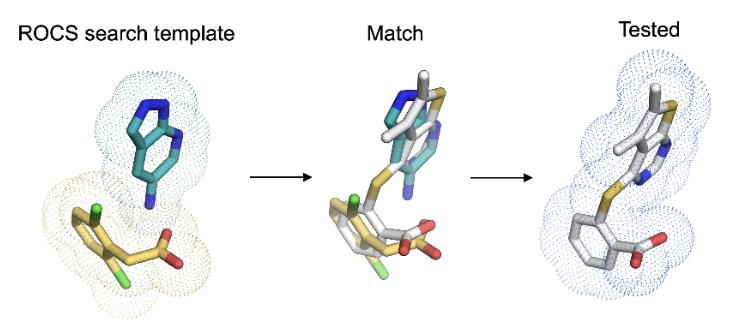
- PMID: 40435250
- PMCID: PMC12118597
- BioRxiv Preprint: 609621
- Full Text
- Deposited Structures: 7HCB, 7HCC, 7HCD, 7HCE, 7HCF, 7HCG, 7HCH, 7HCI, 7HCJ, 7HCK, 7HCL, 7HCM, 7HCN, 7HCO, 7HCP, 7HCQ, 7HCR, 7HCS, 7HCT, 7HCU, 7HCV, 7HCW, 7HCX, 7HCY, 7HCZ, 7HD0, 7HD1, 7HD2, 7HD3, 7HD4, 7HD5, 7HD6, 7HD7, 7HD8, 7HD9, 7HDA, 7HDB, 7HDC, 7HDD, 7HDE, 7HDF, 7HDG, 7HDH, 7HDI, 7HDJ, 7HDK, 7HDL, 7HDM, 7HDN, 7HDO, 7HDP, 7HDQ, 7HDR, 7HDS, 7HDT, 7HDU, 7HDV, 7HDW, 7HDX, 7HDY, 7HDZ, 7HE0, 7HE1, 7HE2, 7HE3, 7HE4, 7HE5, 7HE6, 7HE7, 7HE8, 7HE9, 7HEA, 7HEB, 7HEC, 7HED, 7HEE, 7HEF, 7HEG, 7HEH, 7HEI, 7HEJ, 7HEK, 7HEL, 7HEM, 7HEN, 7HEO, 7HEP, 7HEQ, 7HER, 7HES, 7HET, 7HEU, 7HEV, 7HEW, 7HEX, 7HEY, 7HEZ, 7HF0, 7HF1, 7HF2, 7HF3, 7HF4, 7HF5, 7HF6, 7HF7, 7HF8, 7HF9, 7HFA, 7HFB, 7HFC, 7HFD, 7HFE, 7HFF, 7HFG, 7HFH, 7HFI, 7HFJ, 7HFK, 7HFL, 7HFM, 7HFN, 7HFO, 7HFP, 7HFQ, 7HFR, 7HFS, 7HFT, 7HFU, 7HFV, 7HFW, 7HFX, 7HFY, 7HFZ, 9D6B, 9D6G, 9D6H, 9D6I
- Zenodo Record: 13368547 (PanDDA analysis of ligand screen against the NSP3 macrodomain of SARS-CoV-2: ligands from FrankenROCS fragment-linking pipeline and subsequent optimization of AVI-313)
- GitHub Repository: PatWalters/TS (Thompson Sampling)
- Practical Fragments:Searching monstrously large chemical space with FrankenROCS
- Relay Therapeutics
- QCRG AViDD Program
- Renlso lab @ UCSF
Access the paper
Additional Links
Where to House Big Data on Small Fragments?
Erlanson DA, Burley SK, Fearon D, Fraser JS, Kreitler D, Nonato MC, Sakai N, Wollenhaupt J, Weiss MS
Nature Communications, 2025
- PMID: 40325009
- PMCID: PMC12052810
- ChemRxiv Preprint: 2025-hjjnj
- Full Text
Access the paper
Advances in uncovering the mechanisms of macromolecular conformational entropy
Wankowicz SA, Fraser JS
Nature Chemical Biology, 2025
- PMID: 40275100
- PMCID: PMC12103944
- ChemRxiv Preprint: 2023-9b5k7-v3
- Full Text
- Celebratory tweetstorm from Stephanie Wankowicz
- Wankowicz Lab @ Vanderbilt
Access the paper
Additional Links
MET variants with activating N-lobe mutations identified in hereditary papillary renal cell carcinomas still require ligand stimulation
Guerin C, Vinchent A, Fernandes M, Damour I, Laratte A, Tellier R, Estevam GO, Meneboo JP, Villenet C, Descarpentries C, Fraser JS, Figeac M, Cortot AB, Rouleau E, Tulasne D
Molecular Oncology, 2025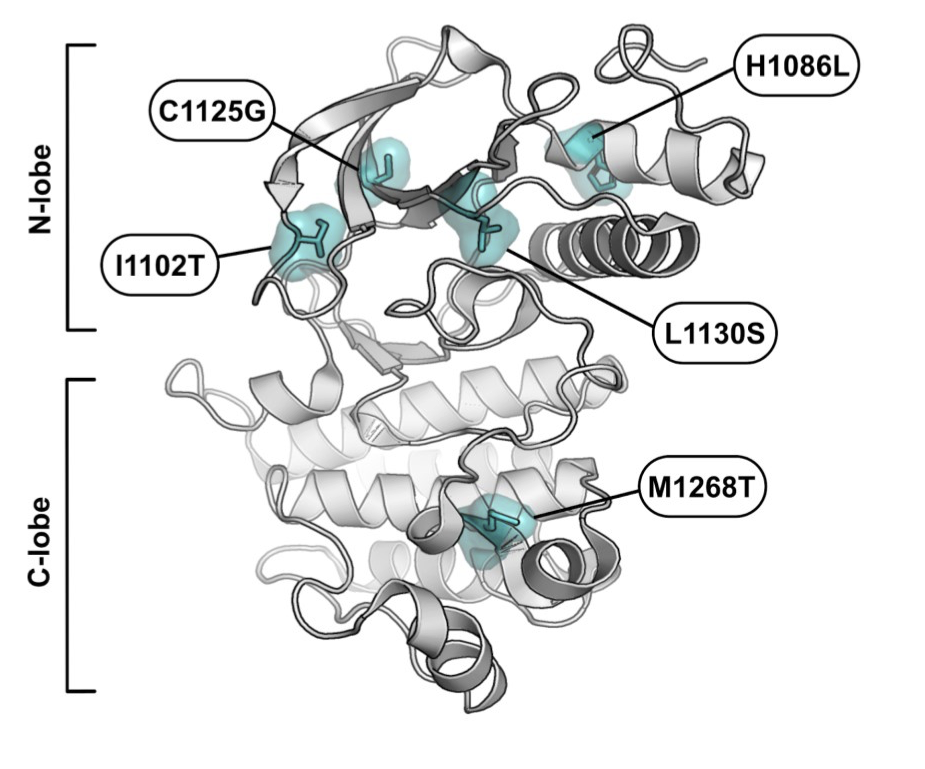
- PMID: 39980226
- PMCID: PMC12330938
- BioRxiv Preprint: 565283
- Full Text
Access the paper
Mapping kinase domain resistance mechanisms for the MET receptor tyrosine kinase via deep mutational scanning
Estevam GO, Linossi EM, Rao J, Macdonald CM, Ravikumar A, Chrispens KM, Capra JA, Coyote-Maestas W, Pimentel H, Collisson EA, Jura N, Fraser JS
eLife, 2025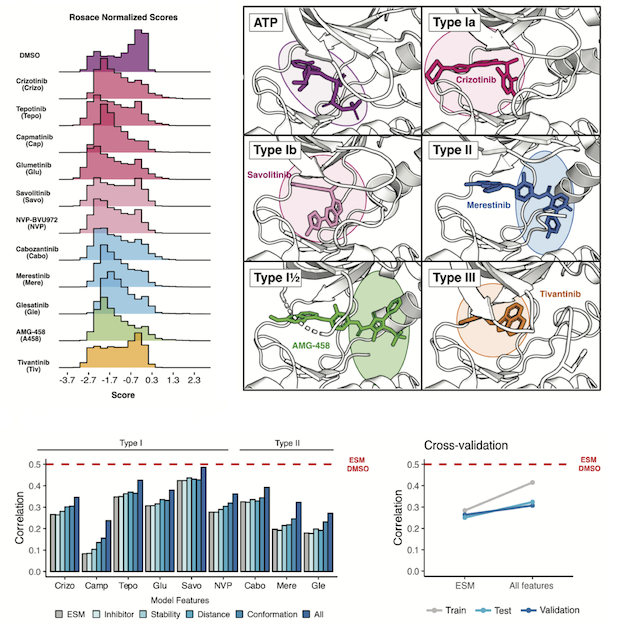
- PMID: 39960754
- PMCID: PMC11832172
- BioRxiv Preprint: 603579
- Full Text
- GitHub Repository: fraser-lab/MET_kinase_Inhibitor_DMS (MET_kinase_Inhibitor_DMS)
- Jura lab @ UC San Francisco
- Collisson lab @ Fred Hutch Cancer Center
- Celebratory Tweetstorm/Xstorm? by Gabriella Estevam
Access the paper
Additional Links
Context-specific inhibition of mitochondrial ribosomes by phenicol and oxazolidinone antibiotics
Bibel B, Raskar T, Couvillion M, Lee M, Kleinman JI, Takeuchi-Tomita N, Churchman LS, Fraser JS, Galonic Fujimori D
Nucleic Acids Research, 2025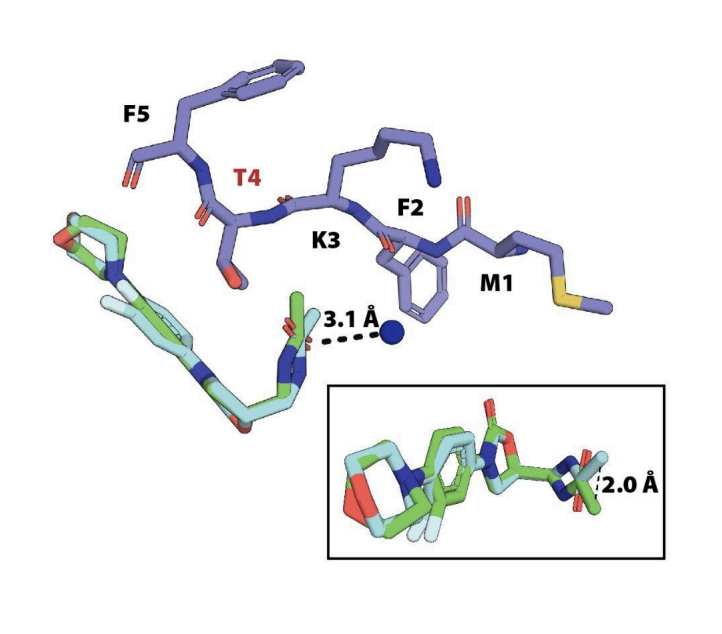
- PMID: 39907106
- PMCID: PMC11795202
- BioRxiv Preprint: 609012
- Full Text
- Deposited Structure and Map: 9CN3/45757
- Churchman lab @ HMS
- Fujimori lab @ UC San Francisco
- Celebratory Tweetstorm/Xstorm? by Brianna Bibel
Access the paper
Additional Links
The impact of Library Size and Scale of Testing on Virtual Screening
Lui F, Mailhot O, Glenn IS, Vigneron SF, Bassim V, Xu X, Valencia KF, Smith MS, Radchenko DS, Fraser JS, Moroz YS, Irwin JJ, Shoichet BK
Nature Chemical Biology, 2025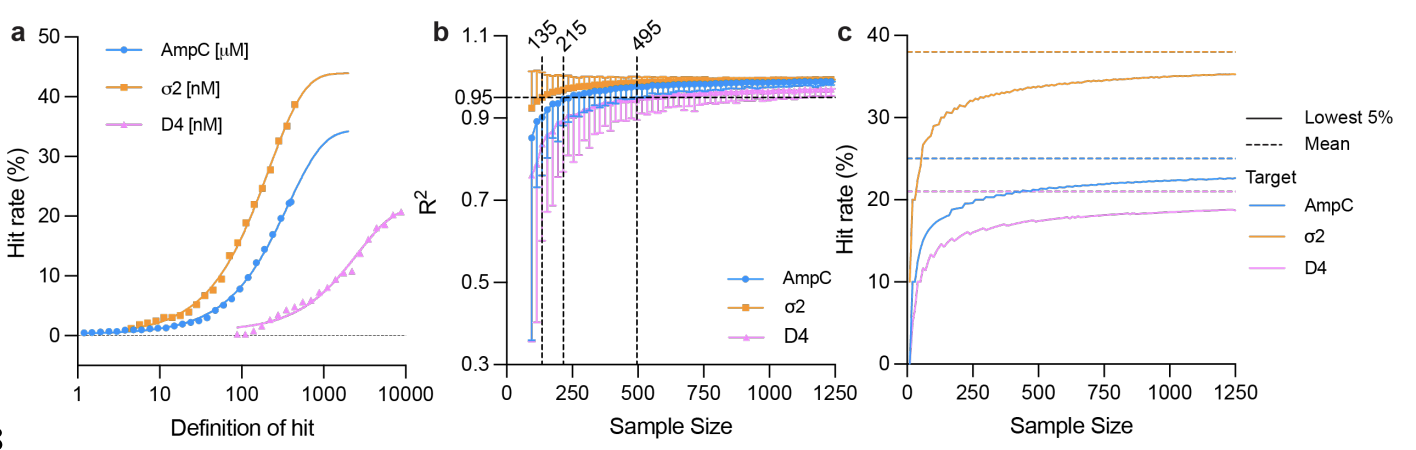
An ILC2-chitinase circuit restores lung homeostasis after epithelial injury
Jung H, Kim DH, Díaz RE, White JM, Rucknagel S, Mosby L, Wang Y, Reddy S, Winkler ES, Hassan AO, Ying B, Diamond MS, Locksley RM, Fraser JS, Van Dyken SJ
Science Immunology, 2024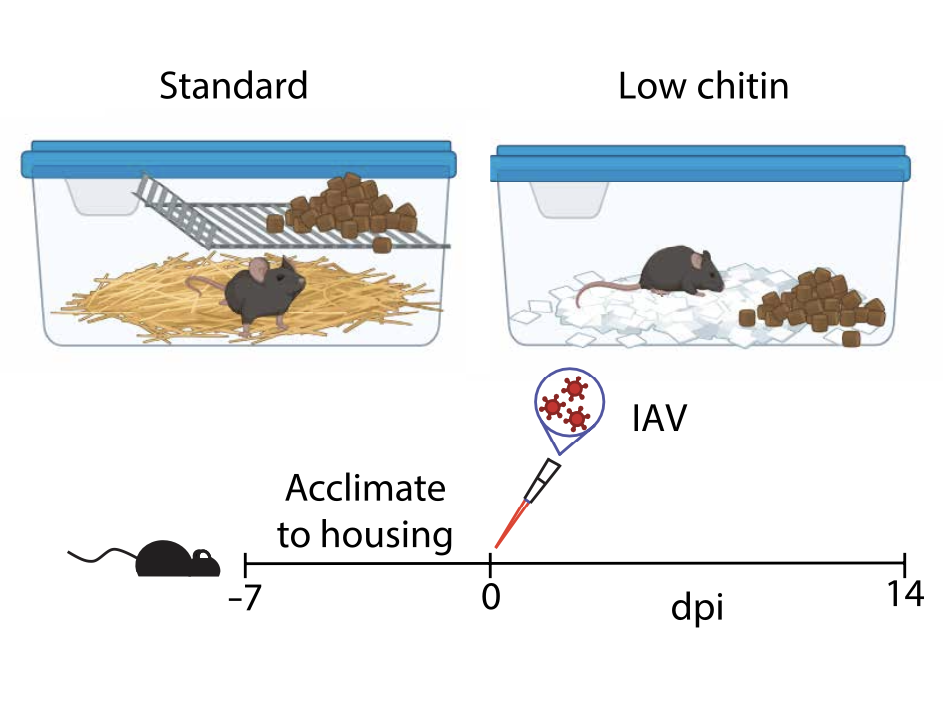
- PMID: 39423283
- PMCID: PMC11854321
- Full Text
- Van Dyken lab @ Washington University in St. Louis
- Locksley lab @ UC San Francisco
Access the paper
Additional Links
A renewed call for open artificial intelligence in biomedicine
Gitter A, Fraser JS, Gonen T, Patro R, Wayment-Steele HK, Williams A, Haibe-Kains B, Dunbrack RL, Cook C, Kundaje A, Hoffman MM, Carpenter AE, Wankowicz SA, Lindorff-Larsen K
Preprint, 2024
- OSF Preprint: 2xh3w
Access the paper
Conserved regulatory motifs in the juxtamembrane domain and kinase N-lobe revealed through deep mutational scanning of the MET receptor tyrosine kinase domain
Estevam GO, Linossi EM, Macdonald CM, Espinoza CA, Michaud JM, Coyote-Maestas W, Collisson EA, Jura N, Fraser JS
eLife, 2024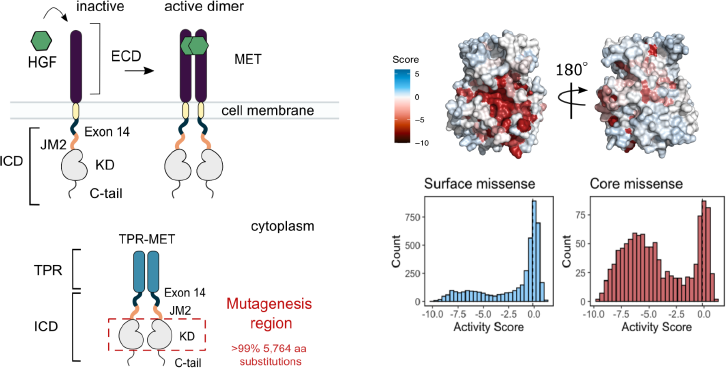
- PMID: 39268701
- PMCID: PMC11398868
- BioRxiv Preprint: 551866
- Full Text
- GitHub Repository: fraser-lab/MET_KinaseDomain_DMS (MET_KinaseDomain_DMS)
- Jura lab @ UC San Francisco
- Collisson lab @ UC San Francisco
- Celebratory Tweetstorm/Xstorm? by Gabriella Estevam
Access the paper
Additional Links
Deep mutational scanning of EccD3 reveals the molecular basis of its essentiality in the mycobacterium ESX secretion system
Trinidad DD, Macdonald CB, Rosenberg OS, Fraser JS, Coyote-Maestas W
Biorxiv, 2024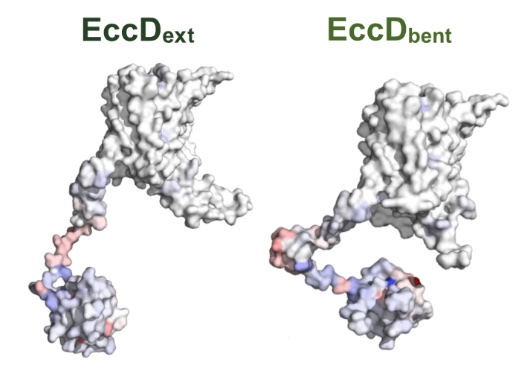
- BioRxiv Preprint: 609456
- GitHub Repository: ddtrini/EccD3_DMS (EccD3_DMS)
- Coyote-Maestas Lab @ UCSF
Access the paper
Additional Link
Ribosomal RNA methylation by GidB modulates discrimination of mischarged tRNA
Bi Z*, Chen YX*, Young ID*, Su HW, Chen Y, Hong JY, Fraser JS, Javid B
Biorxiv, 2024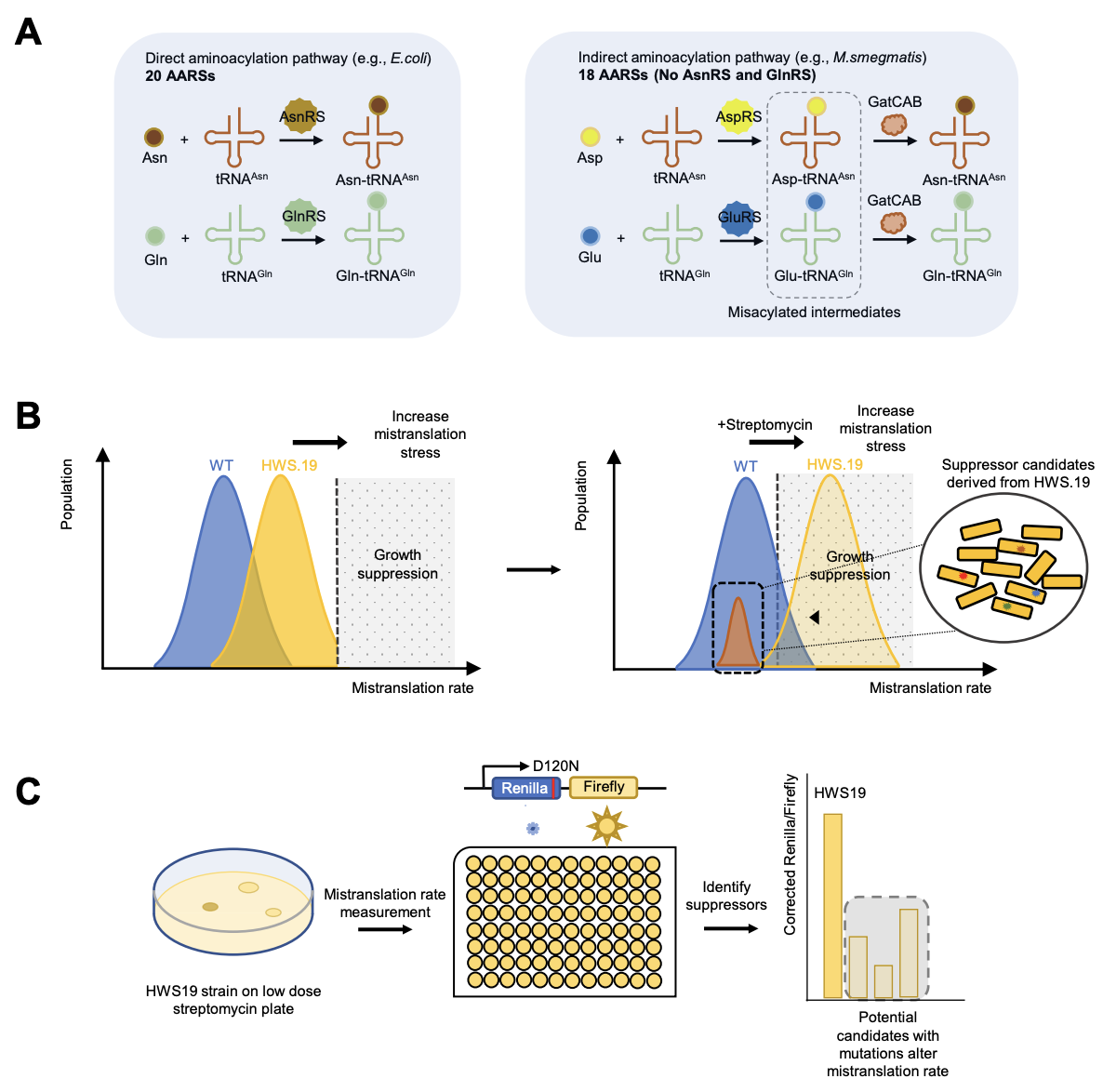
Comprehensive Encoding of Conformational and Compositional Protein Structural Ensembles through mmCIF Data Structure
Wankowicz SA, Fraser JS
IUCrJ, 2024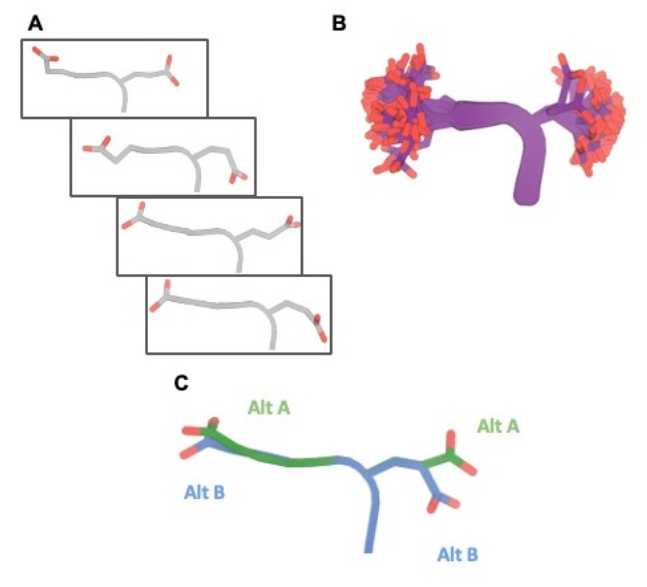
- PMID: 38958015
- PMCID: PMC11220883
- ChemRxiv Preprint: 2023-ggd1w-v3
- Full Text
- Celebratory tweetstorm from Stephanie Wankowicz
Access the paper
Additional Link
Automated Multiconformer Model Building for X-ray Crystallography and Cryo-EM
Wankowicz SA, Ravikumar A, Sharma S, Riley B, Raju A, Flowers J, Hogan DW, van den Bedem H, Keedy DA, Fraser JS
eLife, 2024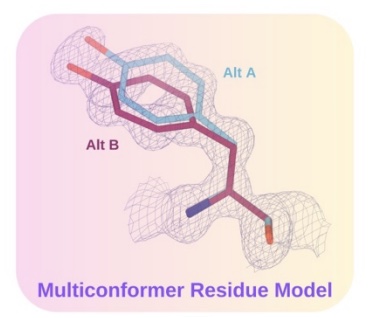
- PMID: 38904665
- PMCID: PMC11192534
- BioRxiv Preprint: 546963
- Full Text
- Zenodo Record: 10936292 (qFit modeled PDBs)
- GitHub Repository: ExcitedStates/qfit-3.0 (qFit)
- Keedy lab @ CUNY Advanced Science Research Center
- Celebratory tweetstorm from Stephanie Wankowicz
Access the paper
Additional Links
Structural characterization of ligand binding and pH-specific enzymatic activity of mouse Acidic Mammalian Chitinase
Díaz RE, Ecker AK, Correy GJ, Asthana P, Young ID, Faust B, Thompson MC, Seiple IB, Van Dyken SJ, Locksley RM, Fraser JS.
eLife, 2024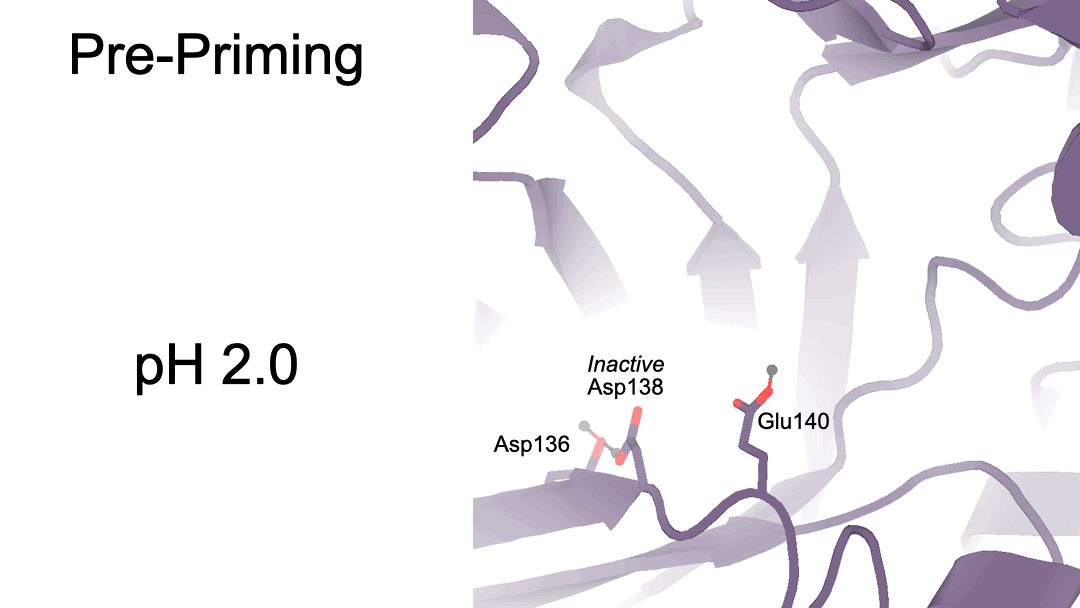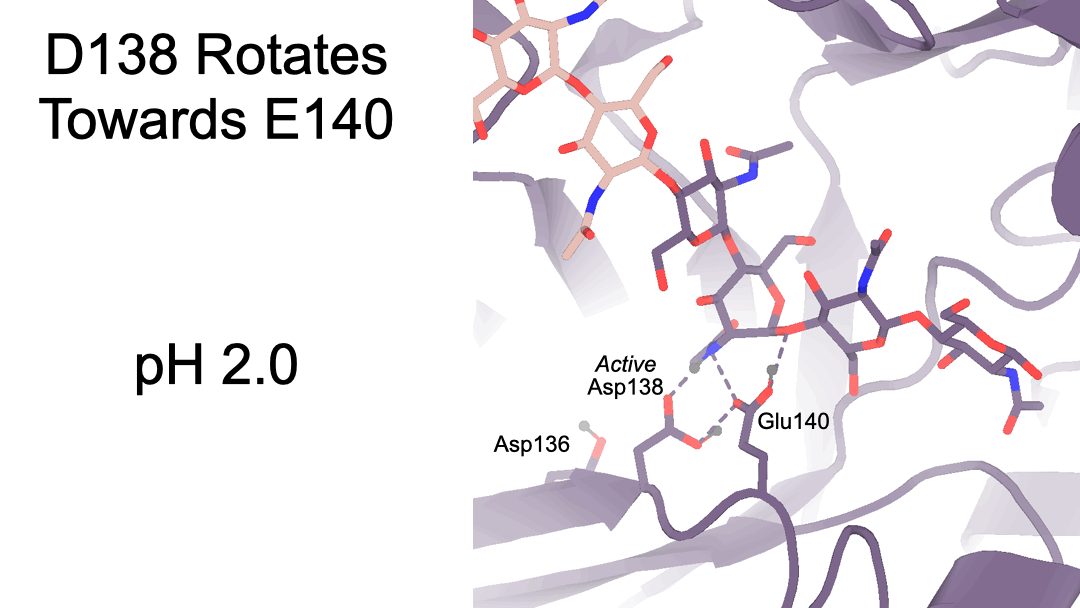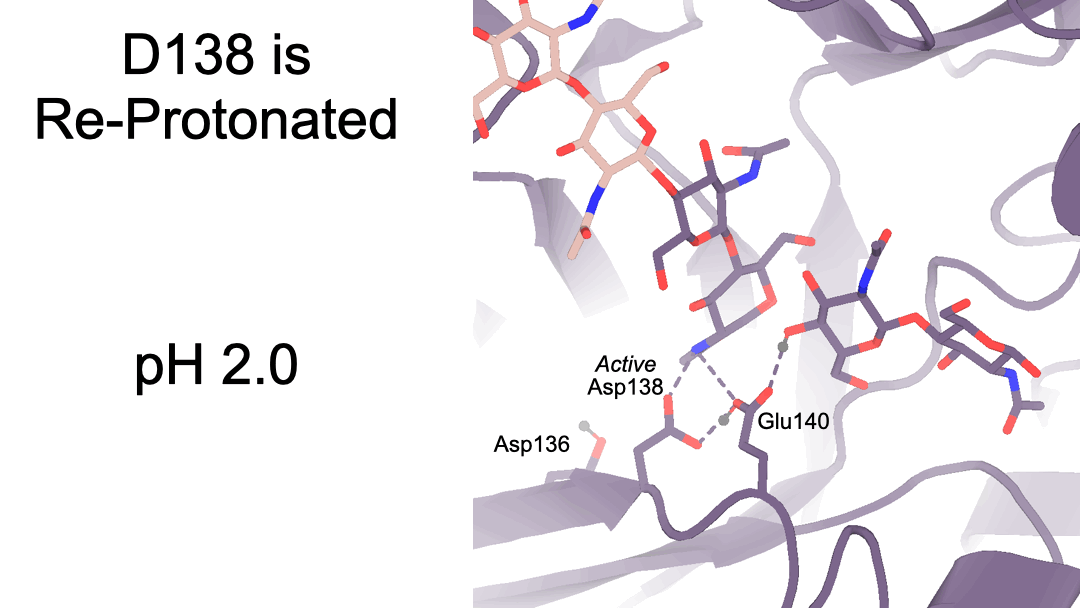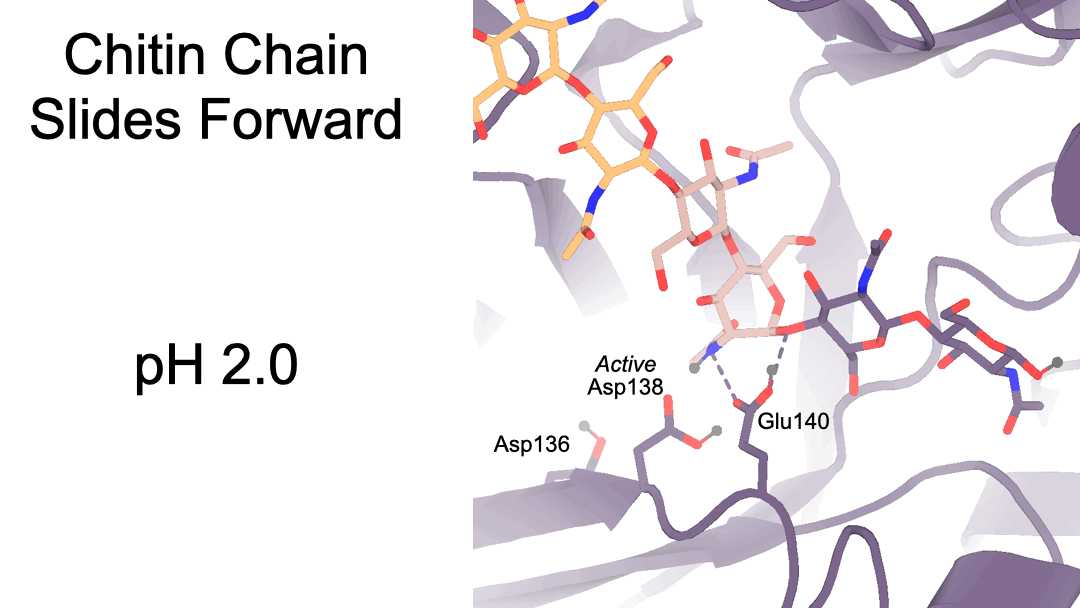
- PMID: 38884443
- PMCID: PMC11182645
- BioRxiv Preprint: 542675
- Full Text
- Deposited Structures: 8FG5, 8FG7, 8GCA, 8FRC, 8FR9, 8FRB, 8FRD, 8FRG, 8FRA
- Zenodo Records: 8250616 (Kinetic properties of mAMCase catalytic domain at various pH.), 7967930 (Characterization of mAMCase sugar-binding subsites.), 7905828 (Asp138 orientation correlates with ligand subsite occupancy.), 7905863 (pKa of GH18 chitinases in the inactive and active conformation.), 7758821 (10 ns Molecular Dynamics simulations of mAMCase at pH 2.0 and 6.5 in complex with GlcNAc6.), 7967958 (Proposed catalytic mechanism of mAMCase.), 7905944 (Crystallization conditions of apo and holo mAMCase.), 7967978 (Overview of key residues for mAMCase activity.), 7967954 (Protein-ligand interactions between mAMCase and chitin.), 7758815 (Ringer analysis of catalytic triad confirms alternative Asp138 conformations.)
- Addgene Plasmids: 200229 (mAMCase Catalytic Domain in a pcDNA3.1 vector), 200228 (mAMCase Catalytic Domain in a pTwist CMV vector)
- Thompson lab @ UC Merced
- Seiple lab @ UC San Francisco
- Van Dyken lab @ Washington University in St. Louis
- Locksley lab @ UC San Francisco
Access the paper
Additional Links
Rosace: a robust deep mutational scanning analysis framework employing position and mean-variance shrinkage
Rao J, Xin R, Macdonald CB, Howard M, Estevam GO, Yee SW, Wang M, Fraser JS, Coyote-Maestas W, Pimentel H
Genome Biology, 2024
- PMID: 38789982
- PMCID: PMC11127319
- BioRxiv Preprint: 562292
- Full Text
- GitHub Repository: pimentellab/rosace (Github repository for Rosace)
- Tweetstorm by corresponding author Jingyou Rao
- Coyote-Maestas Lab
- Pimentel Lab
Access the paper
Additional Links
AlphaFold3 Transparency and Reproducibility
Wankowicz SA, Beltrao P, Cravatt B, Dunbrack R, Gitter A, Lindorff-Larsen K, Ovchinnikov S, Polizzi N, Shoichet B, Fraser JS
Preprint, 2024
- Zenodo Record: 11391920 (Open letter and endorsement list)
- AlphaFold3 Validation and the Role of Journals
- Retraction Watch - "Nature earns ire over lack of code availability for Google DeepMind protein folding paper"
- Science - "ScienceInsider: Limits on access to DeepMind’s new protein program trigger backlash"
- C&EN - "AlphaFold 3 to offer structure prediction via web browser"
- Nature Editorial - "AlphaFold3 — why did Nature publish it without its code?"
- Nature News - "Who will make AlphaFold3 open source? Scientists race to crack AI model"
- Undark - Why AlphaFold 3 Needs to Be Open Source
- GEN/EDGE - AlphaFold 3 Angst: Limited Accessibility Stirs Outcry from Researchers
- Science - "ScienceInsider: Google DeepMind releases code behind its most advanced protein prediction program"
Access the paper
Additional Links
Recommendations for accelerating open preprint peer review to improve the culture of science
Avissar-Whiting M, Belliard F, Bertozzi SM, Brand A, Brown K, Clément-Stoneham G, Dawson S, Dey G, Ecer D, Edmunds SC, Farley A, Fischer TD, Franko M, Fraser JS, Funk K, Ganier C, Harrison M, Hatch A, Hazlett H, Hindle S, Hook DW, Hurst P, Kamoun S, Kiley R, Lacy MM, LaFlamme M, Lawrence R, Lemberger T, Leptin M, Lumb E, MacCallum CJ, Marcum CS, Marinello G, Mendonça A, Monaco S, Neves K, Pattinson D, Polka JK, Puebla I, Rittman M, Royle SJ, Saderi D, Sever R, Shearer K, Spiro JE, Stern B, Taraborelli D, Vale R, Vasquez CG, Waltman L, Watt FM, Weinberg ZY, Williams M.
PLOS Biology, 2024
- PMID: 38421949
- PMCID: PMC10903809
- OSF Preprint: cht8p
- Recognizing Preprint Peer Review meeting at Janelia
- James's keynote from the meeting
Access the paper
Additional Links
Structure is beauty, but not always truth
Fraser JS, Murcko MA
Cell, 2024
- PMID: 38306978
- PMCID: PMC10947451
- Full Text
- In the pipeline: John Keats Would Like a Word
Access the paper
Additional Link
Discovery and clinical proof-of-concept of RLY-2608, a first-in-class mutant-selective allosteric PI3Ka inhibitor that decouples anti-tumor activity from hyperinsulinemia
Varkaris A, Pazolli E, Gunaydin H, Wang Q, Pierce L, Boezio AA, Bulku A, DiPietro L, Fridrich C, Frost A, Giordanetto F, Hamilton EP, Harris K, Holliday M, Hunter TL, Iskandar A, Ji Y, Larivée A, LaRochelle JR, Lescarbeau A, Llambi F, Lormil B, Mader MM, Mar BG, Martin I, McLean TH, Michelsen K, Pechersky Y, Puente-Poushnejad E, Raynor K, Rogala D, Samadani R, Schram AM, Shortsleeves K, Swaminathan S, Tajmir S, Tan G, Tang Y, Valverde R, Wehrenberg B, Wilbur J, Williams BR, Zeng H, Zhang H, Walters WP, Wolf BB, Shaw DE, Bergstrom DA, Watters J, Fraser JS, Fortin PD, Kipp DR.
Cancer Discovery, 2023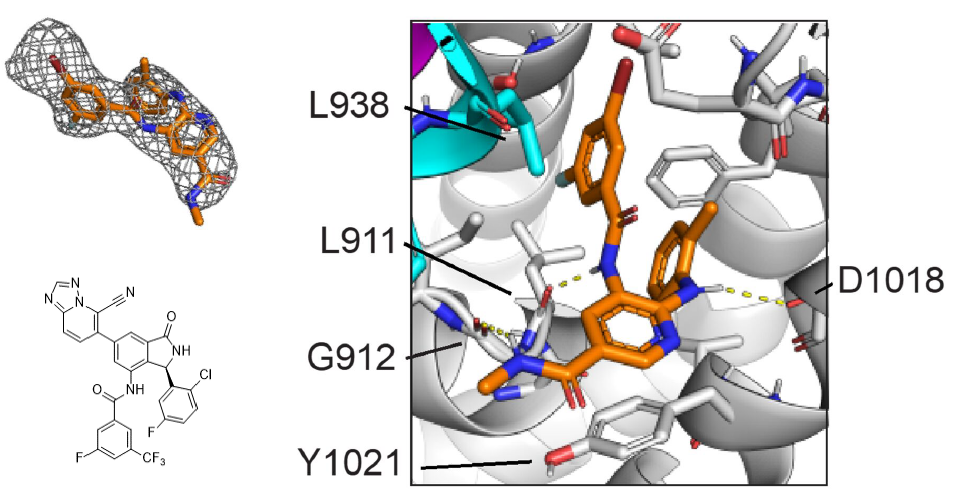
- PMID: 37916956
- PMCID: PMC10850943
- Full Text
- Deposited Structures: 8TU6, 8TS8, 8TS7, 8TSB, 8TSA, 8TS9, 8TSC, 8TSD
- Relay Therapeutics
- D.E. Shaw Research
- Precision Targeting of Mutant PI3Kα
- A New Wave of PI3Kα Inhibitors
- Allosteric PI3Kα Inhibition Overcomes On-target Resistance to Orthosteric Inhibitors Mediated by Secondary PIK3CA Mutations
Access the paper
Additional Links
Mapping Protein Dynamics at High-Resolution with Temperature-Jump X-ray Crystallography
Wolff AM, Nango E, Young ID, Brewster AS, Kubo M, Nomura T, Sugahara M, Owada S, Barad BA, Ito K, Bhowmick A, Carbajo S, Hino T, Holton JM, Im D, O’Riordan LJ, Tanaka T, Tanaka R, Sierra RG, Yumoto F, Tono K, Iwata S, Sauter NK, Fraser JS, Thompson MC
Nature Chemistry, 2023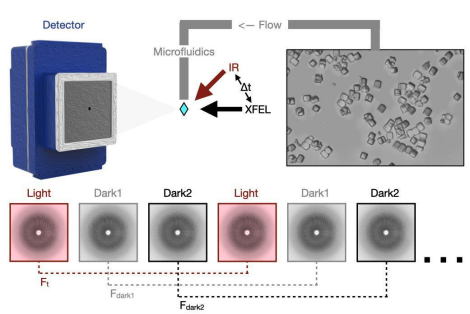
- PMID: 37723259
- PMCID: PMC10624634
- BioRxiv Preprint: 495662
- Full Text
- Deposited Structures: 8CVU, 8CVV, 8CVW, 8CW0, 8CW1, 8CW3, 8CW5, 8CW6, 8CW7, 8CW8, 8CWB, 8CWC, 8CWD, 8CWE, 8CWF, 8CWG, 8CWH
- GitHub Repository: mthompson-lab/Temperature-Jump-Crystallography_Analysis-for-Paper
- Nango lab @ Tohoku University
- Thompson lab @ UC Merced
- BioSciences @ Lawrence Berkeley National Laboratory
- Celebratory Tweetstorm by senior author Mike Thompson
- In the Pipeline - Warming Proteins Up Under the X-ray Beam
Access the paper
Additional Links
A type 2 immune circuit in the stomach controls mammalian adaptation to dietary chitin
Kim D, Wang Y, Jung H, Field RL, Zhang X, Liu T, Ma C, Fraser JS, Brestoff JR, Van Dyken SJ
Science, 2023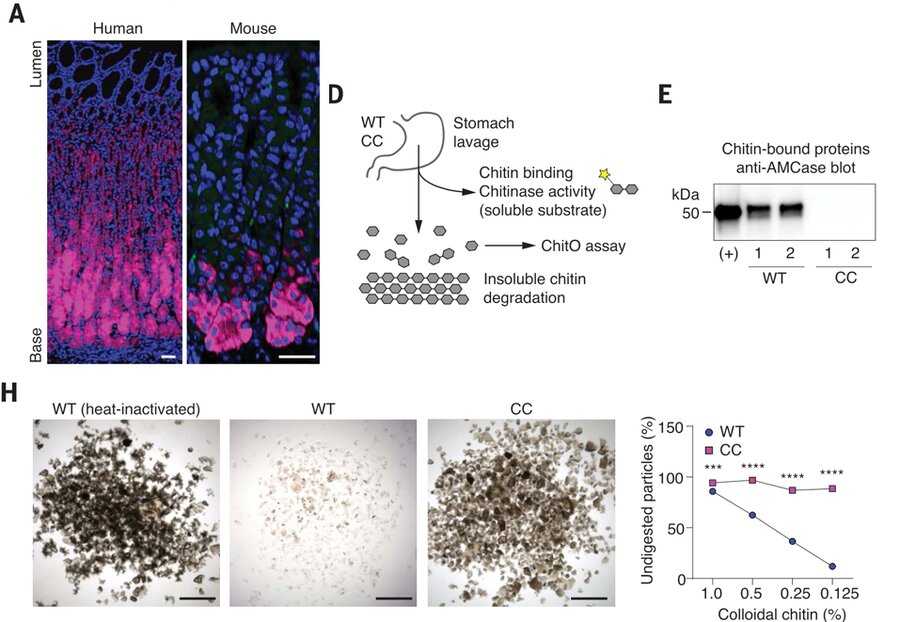
- PMID: 37676935
- PMCID: PMC10624634
- Full Text
- Van Dyken Lab @ WUSTL
Access the paper
Additional Link
A single inactivating amino acid change in the SARS-CoV-2 NSP3 Mac1 domain attenuates viral replication and pathogenesis in vivo
Taha TY*, Suryawanshi RK*, Chen IP*, Correy GJ*, O’Leary PC, Jogalekar MP, McCavitt-Malvido M, Diolaiti M, Kimmerly GR, Tsou CL, Martinez-Sobrido L, Krogan NJ, Ashworth A, Fraser JS, Ott M
PLoS Pathogens, 2023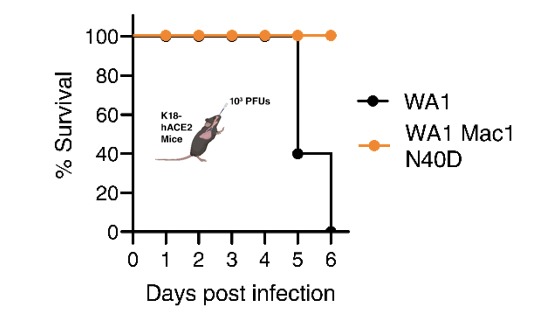
- PMID: 37651466
- PMCID: PMC10499221
- BioRxiv Preprint: 537104
- Full Text
- Deposited Structures: 8SH6, 8SH8
- Celebratory tweetstorm from James Fraser
- Ott lab @ Gladstone
- Alan Ashworth @ UCSF
Access the paper
Additional Links
Chemoenzymatic syntheses of fluorine-18-labeled disaccharides from [18F]FDG yield potent sensors of living bacteria in vivo
Sorlin A, Alvarez ML, Rabbitt S, Alanizi A, Shuere B, Bobba KN, Blecha J, Sakhamuri S, Evans M, Bayles KJ, Flavell R, Rosenberg O, Sriram R, Desmet T, Nidetzky B, Engel J, Ohliger M, Fraser JS, Wilson DM
JACS, 2023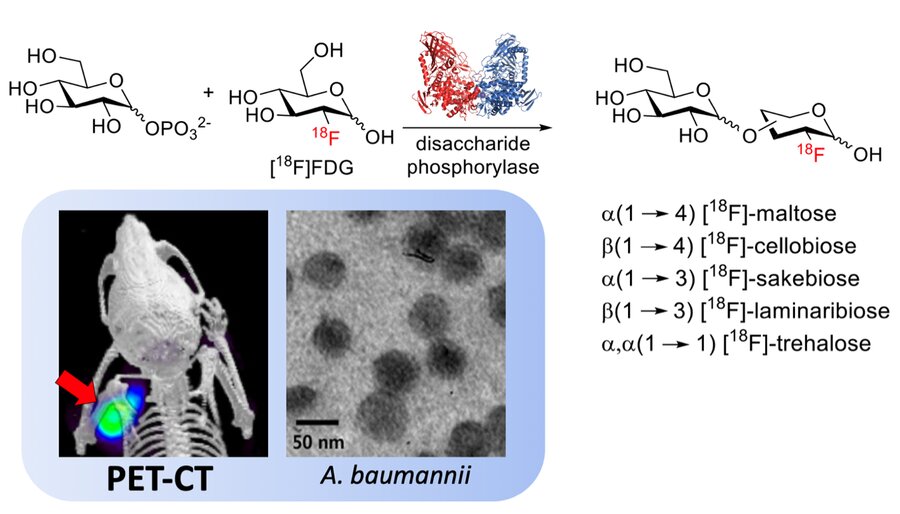
- PMID: 37535945
- PMCID: PMC10436271
- BioRxiv Preprint: 541529
- Full Text
- Wilson Lab @ UCSF
Access the paper
Additional Link
Refinement of Multiconformer Ensemble Models from Multi-temperature X-ray Diffraction Data
Du S, Wankowicz SA, Yabukarski F, Doukov T, Herschlag D, Fraser JS
Methods in Enzymology, 2023
- PMID: 37748828
- PMCID: PMC10637719
- BioRxiv Preprint: 539620
- Full Text
- Deposited Structures: 8SOG, 8SQV, 8SPL, 8SOV, 8SOU
- Herschlag lab @ Stanford University
- Celebratory tweetstorm from Stephanie Wankowicz
Access the paper
Additional Links
Molecular evidence of widespread benzimidazole drug resistance in Ancylostoma caninum from domestic dogs throughout the USA and discovery of a novel isotype-1 β-tubulin benzimidazole resistance mutation
Venkatesan A, Jimenez Castro PD, Morosetti A, Horvath H, Chen R, Redman E, Dunn K, Collins JB, Fraser JS, Andersen EC, Kaplan RM, Gilleard JS
PLoS Pathogens, 2023
- PMID: 36862759
- PMCID: PMC10013918
- BioRxiv Preprint: 511840
- Full Text
- GitHub Repository: AndersenLab/2022_BZ_Resistance_ben1_Q134H (Benzimidazole resistance growth measurements)
- Andersen lab @ Northwestern University
- Gilleard lab @ University of Calgary
- Celebratory Tweetstorm by corresponding author John Gilleard
- Gilleard lab @ University of Calgary
Access the paper
Additional Links
Deep insertion, deletion, and missense mutation libraries for exploring protein variation in evolution, disease, and biology
Macdonald CB, Nedrud D, Rockefeller Grimes P, Trinidad D, Fraser JS, Coyote-Maestas W
Genome Biology, 2023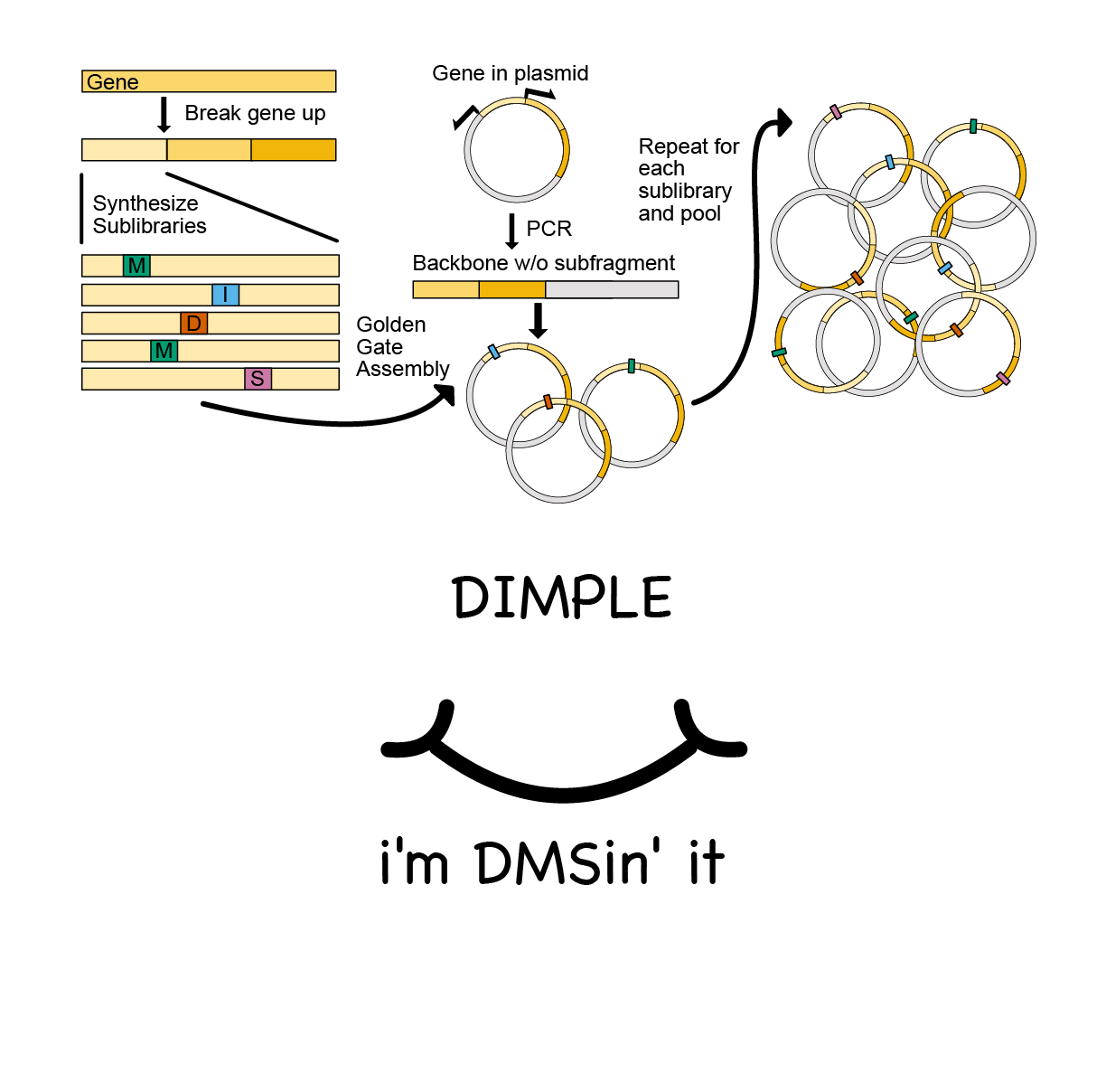
- PMID: 36829241
- PMCID: PMC9951526
- BioRxiv Preprint: 501589
- Full Text
- GitHub Repository: coywil26/DIMPLE (DIMPLE)
- Celebratory Tweetstorm by Willow Coyote-Maestas
Access the paper
Additional Link
Bacteriophages inhibit and evade cGAS-like immune function in bacteria
Huiting E*, Cao X*, Ren J, Athukoralage JS, Luo Z, Silas S, An N, Carion H, Zhou Y, Fraser JS, Feng Y, Bondy-Denomy J
Cell, 2023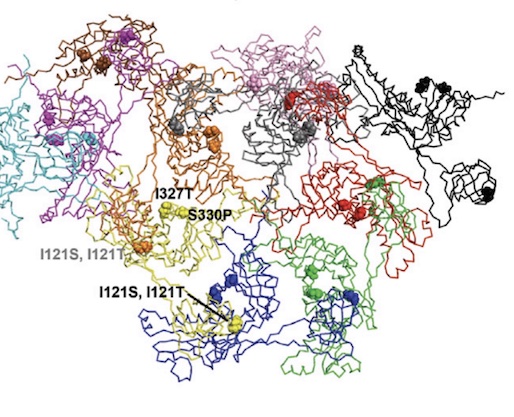
- PMID: 36750095
- PMCID: PMC9975087
- BioRxiv Preprint: 486325
- Full Text
- Deposited Structures: 8H2X, 8H2J, 8H39
- Bondy-Denomy lab @ UC San Francisco
- Celebratory Tweetstorm from Erin Huiting
Access the paper
Additional Links
Deep neural language modeling enables functional protein generation across families
Madani A, Krause B*, Greene ER*, Subramanian S, Mohr BP, Holton JM, Olmos Jr. JL, Xiong C, Sun ZZ, Socher R, Fraser JS, Naik N
Nature Biotechnology, 2023
- PMID: 36702895
- PMCID: PMC10400306
- BioRxiv Preprint: 452833
- Full Text
- Deposited Structure: 7RGR
- Salesforce Research
- Learning from evolution
- Celebratory Tweetstorm by Ali Madani
- Celebratory Tweetstorm by James Fraser
- News and Views - Protein engineering - Hallucinating functional protein sequences
- Nature Biotechnology Editorial - Generating ‘smarter’ biotechnology
- Nature Biotechnology News - AI-enhanced protein design makes proteins that have never existed
- In the Pipeline - Making Up Proteins
- New Scientist - AI has designed bacteria-killing proteins from scratch – and they work
- Vice Motherboard - AI Replicated Evolution and Generated New Enzymes as Good as Natural Ones
- IEEE Spectrum - GPT Protein Models Speak Fluent Biology
- BioIT World - Natural Language Processing Designs Original Proteins From Scratch
- GEN Biotechnology - Learning to Read and Write in the Language of Proteins
- Advanced Light Source - Chatbot-Style AI Designs Novel Functional Protein
- UCSF - AI Technology Generates Original Proteins from Scratch
Access the paper
Additional Links
Iterative computational design and crystallographic screening identifies potent inhibitors targeting the Nsp3 Macrodomain of SARS-CoV-2
Gahbauer S*, Correy GJ*, Schuller M, Ferla MP, Doruk YU, Rachman M, Wu T, Diolaiti M, Wang S, Neitz RJ, Fearon D, Radchenko DS, Moroz YS, Irwin JJ, Renslo AR, Taylor JC, Gestwicki JE, von Delft F, Ashworth A, Ahel I, Shoichet BK, Fraser JS
PNAS, 2023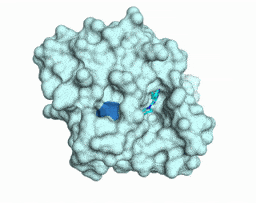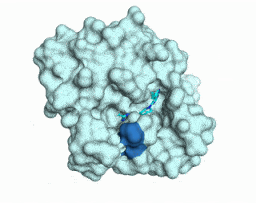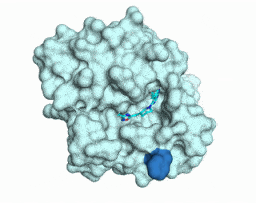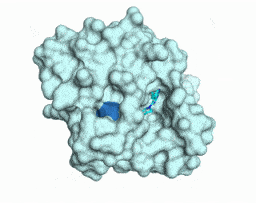
- PMID: 36598939
- PMCID: PMC9926234
- BioRxiv Preprint: 497816
- Full Text
- Deposited Structures: 5SOI, 5SOJ, 5SOK, 5SOL, 5SOM, 5SON, 5SOO, 5SOP, 5SOQ, 5SOR, 5SOS, 5SOT, 5SOU, 5SOV, 5SOW, 5SOX, 5SOY, 5SOZ, 5SP0, 5SP1, 5SP2, 5SP3, 5SP4, 5SP6, 5SP7, 5SP8, 5SP9, 5SPA, 5SPB, 5SPC, 5SPD, 5SPE, 5SPF, 5SPG, 5SPH, 5SPI, 5SPJ, 5SPK, 5SPL, 5SPM, 5SPN, 5SPO, 5SPP, 5SPQ, 5SPR, 5SPS, 5SPT, 5SPU, 5SPV, 5SPW, 5SPX, 5SPY, 5SPZ, 5SQ0, 5SQ1, 5SQ2, 5SQ3, 5SQ4, 5SQ5, 5SQ6, 5SQ7, 5SQ8, 5SQ9, 5SQA, 5SQB, 5SQC, 5SQD, 5SQE, 5SQF, 5SQG, 5SQH, 5SQI, 5SQJ, 5SQK, 5SQL, 5SQM, 5SQN, 5SQO, 5SQP, 5SQQ, 5SQR, 5SQS, 5SQT, 5SQU, 5SQV, 5SQW, 5SQX, 5SQY, 5SQZ, 5SR0, 5SR1, 5SR2, 5SR3, 5SR4, 5SR5, 5SR6, 5SR7, 5SR8, 5SR9, 5SRA, 5SRB, 5SRC, 5SRD, 5SRE, 5SRF, 5SRG, 5SRH, 5SRI, 5SRJ, 5SRK, 5SRL, 5SRM, 5SRN, 5SRO, 5SRP, 5SRQ, 5SRR, 5SRS, 5SRT, 5SRU, 5SRV, 5SRW, 5SRX, 5SRY, 5SRZ, 5SS0, 5SS1, 5SS2, 5SS3, 5SS4, 5SS5, 5SS6, 5SS7, 5SS8, 5SS9, 5SSA, 5SSB, 5SSC, 5SSD, 5SSE, 5SSF, 5SSG, 5SSH, 5SSI, 5SSJ, 5SSK, 5SSL, 5SSM, 5SSN, 5SSO, 5SSP, 5SSQ, 5SSR
- Zenodo Record: 6688239 (Scaled and merged structure factor intensities for all datasets, and the output of Dimple and PanDDA)
- Gestwicki lab @ UC San Francisco
- von Delft lab @ University of Oxford
- Ashworth lab @ UC San Francisco
- Ahel lab @ University of Oxford
- Shoichet lab @ UC San Francisco
- Celebratory Tweetstorm by Stefan Gahbauer
- Celebratory Tweetstorm by James Fraser
- Practical Fragments post on the paper
Access the paper
Additional Links
Molecular-Dynamics simulation methods for macromolecular crystallography
Wych DC, Aoto PC, Vu L, Wolff AM, Mobley DL, Fraser JS, Taylor SS, Wall ME
Acta Crystallographica D, 2023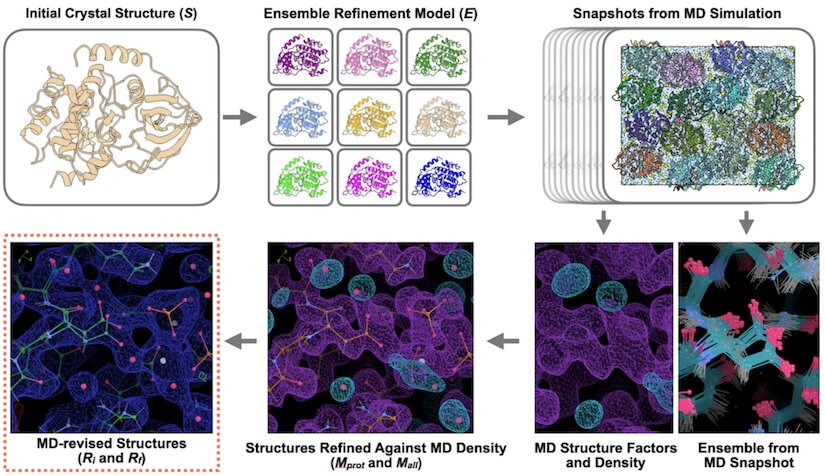
- PMID: 36601807
- PMCID: PMC9926234
- BioRxiv Preprint: 486986
- Full Text
- GitHub Repository: lanl/lunus (MD Snapshots)
- Celebratory Tweetstorm by David C. Wych
Access the paper
Additional Link
Fragment-based hit discovery via unsupervised learning of fragment-protein complexes
McCorkindale W, Ahel I, Barr H, Correy GJ, Fraser JS, London N, Schuller M, Shurrush K, Lee AA
Biorxiv, 2022
- BioRxiv Preprint: 517375
- Deposited Structures: 7FR0, 7FR1, 7FR2, 7FR3, 7FR4, 7FR5, 7FR6, 7FR7, 7FR8, 7FR9, 7FRA, 7FRB, 7FRC, 7FRD
- GitHub Repository: wjm41/fresco (Fragment Ensemble Scoring)
- London lab @ Weizmann Institute
- Lee lab @ University of Cambridge
- Celebratory Tweetstorm from Alpha A. Lee
Access the paper
Additional Links
Ensemble-function relationships to dissect mechanisms of enzyme catalysis
Yabukarski F, Doukov T, Pinney MM, Biel JT, Fraser JS, Herschlag D
Science Advances, 2022
- PMID: 36240280
- PMCID: PMC9565801
- BioRxiv Preprint: 461692
- Full Text
- Deposited Structures: 7RXK, 7RXF, 7RY4
- Pinney lab @ UC San Francisco
- Herschlag lab @ Stanford University
Access the paper
Additional Links
Type 2 innate immunity regulates hair follicle homeostasis to control Demodex pathosymbionts
Ricardo-Gonzalez RR, Kotas ME, Tenvooren I, Marquez DM, Fassett MS, Lee J, Daniel SG, Bittinger K, Díaz RE, Fraser JS, Ansel KM, Spitzer MH, Liang HE, Locksley RM
Immunity, 2022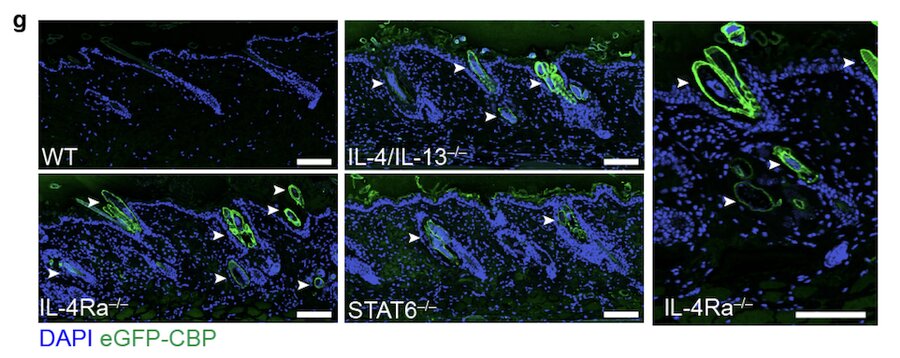
- PMID: 36044899
- PMCID: PMC9561030
- BioRxiv Preprint: 437438
- Full Text
- Ansel lab @ UC San Francisco
- Spitzer lab @ UC San Francisco
- Locksley lab @ UC San Francisco
Access the paper
Additional Links
Of problems and opportunities-How to treat and how to not treat crystallographic fragment screening data
Weiss MS, Wollenhaupt J, Correy GJ, Fraser JS, Heine A, Klebe G, Krojer T, Thunissen M, Pearce NM
Protein Science, 2022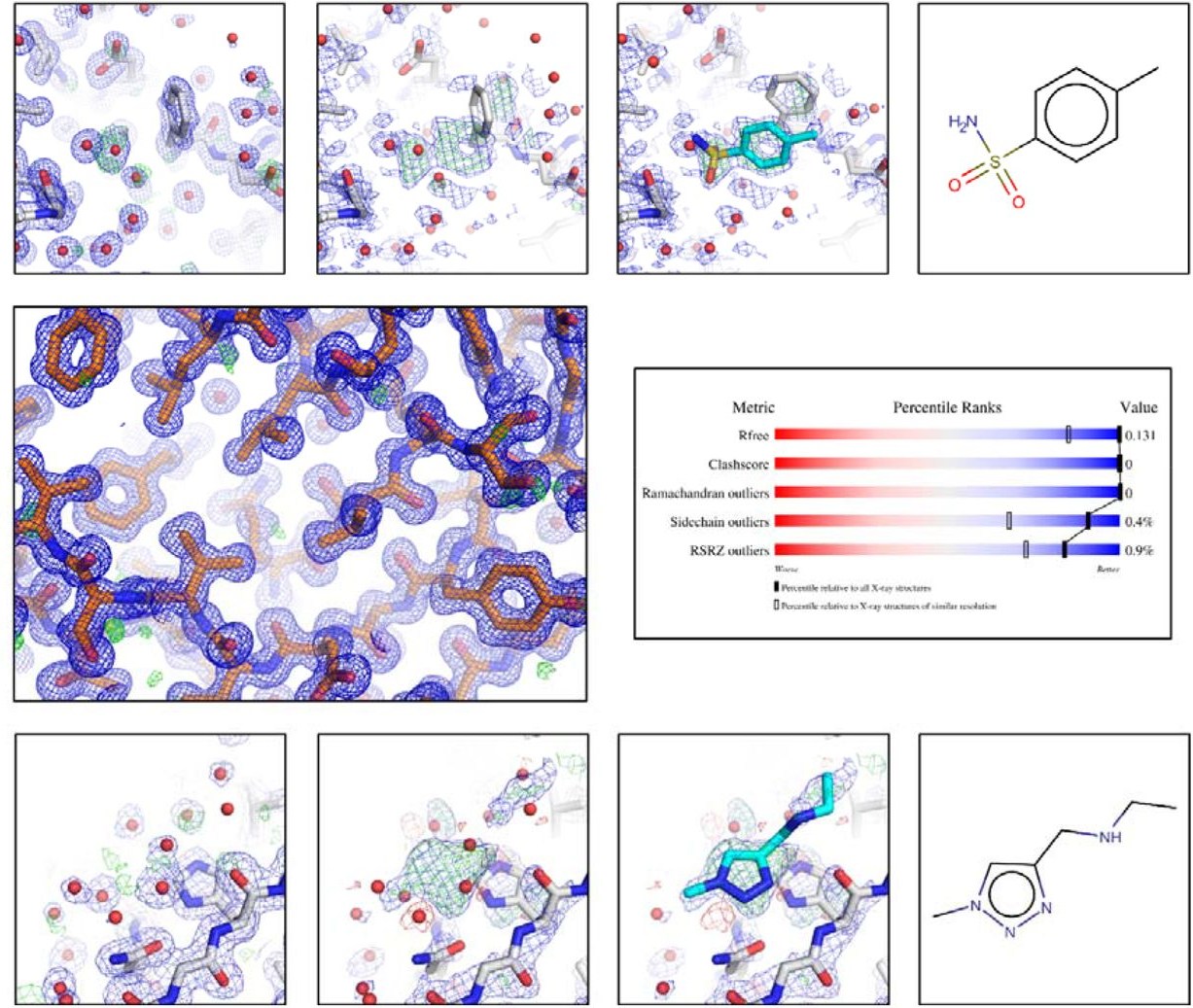
- PMID: 36040268
- PMCID: PMC9424839
- BioRxiv Preprint: 492756
- Full Text
- Structural Genomics Consortium
- MAX IV lab @ Lund University
Access the paper
Additional Links
The mechanisms of catalysis and ligand binding for the SARS-CoV-2 NSP3 macrodomain from neutron and X-ray diffraction at room temperature
Correy GJ, Kneller DW, Phillips G, Pant S, Russi S, Cohen AE, Meigs G, Holton JM, Gahbauer S, Thompson MC, Ashworth A, Coates L, Kovalevsky A, Meilleur F, Fraser JS
Science Advances, 2022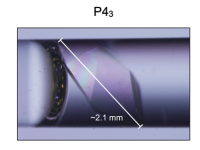
- PMID: 35622909
- PMCID: PMC9140965
- BioRxiv Preprint: 479477
- Full Text
- Deposited Structures: 7TWF, 7TWG, 7TWH, 7TWI, 7TWJ, 7TWN, 7TWO, 7TWP, 7TWQ, 7TWR, 7TWS, 7TWT, 7TWV, 7TWW, 7TWX, 7TWY, 7TX0, 7TX1, 7TX3, 7TX4, 7TX5
- Thompson lab @ UC Merced
- Ashworth lab @ UC San Francisco
- Oak Ridge National Laboratory
- Meilleur lab @ North Carolina State University
- Celebratory Tweetstorm by James Fraser
Access the paper
Additional Links
Ligand binding remodels protein side chain conformational heterogeneity
Wankowicz SA, de Oliveira SHP, Hogan DW, van den Bedem H, Fraser JS
eLife, 2022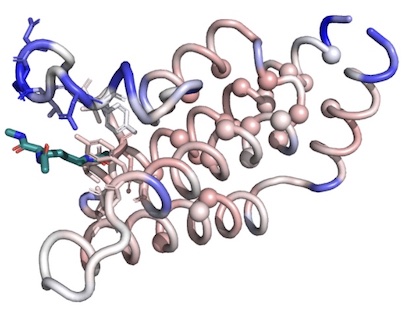
- PMID: 35312477
- PMCID: PMC9084896
- BioRxiv Preprint: 461269
- Full Text
- GitHub Repositories: ExcitedStates/qfit-3.0, fraser-lab/Apo_Holo_Analysis
- Celebratory Tweetstorm by Stephanie Wankowicz
- Blog Post: So you want to do a structural bioinformatic analysis…
Access the paper
Additional Links
Accurate positioning of functional residues with robotics-inspired computational protein design
Krivacic C*, Kundert K*, Pan X*, Pache RA*, Liu L, Conchúir SO, Jeliazkov JR, Gray JJ, Thompson MC, Fraser JS, Kortemme T
PNAS, 2022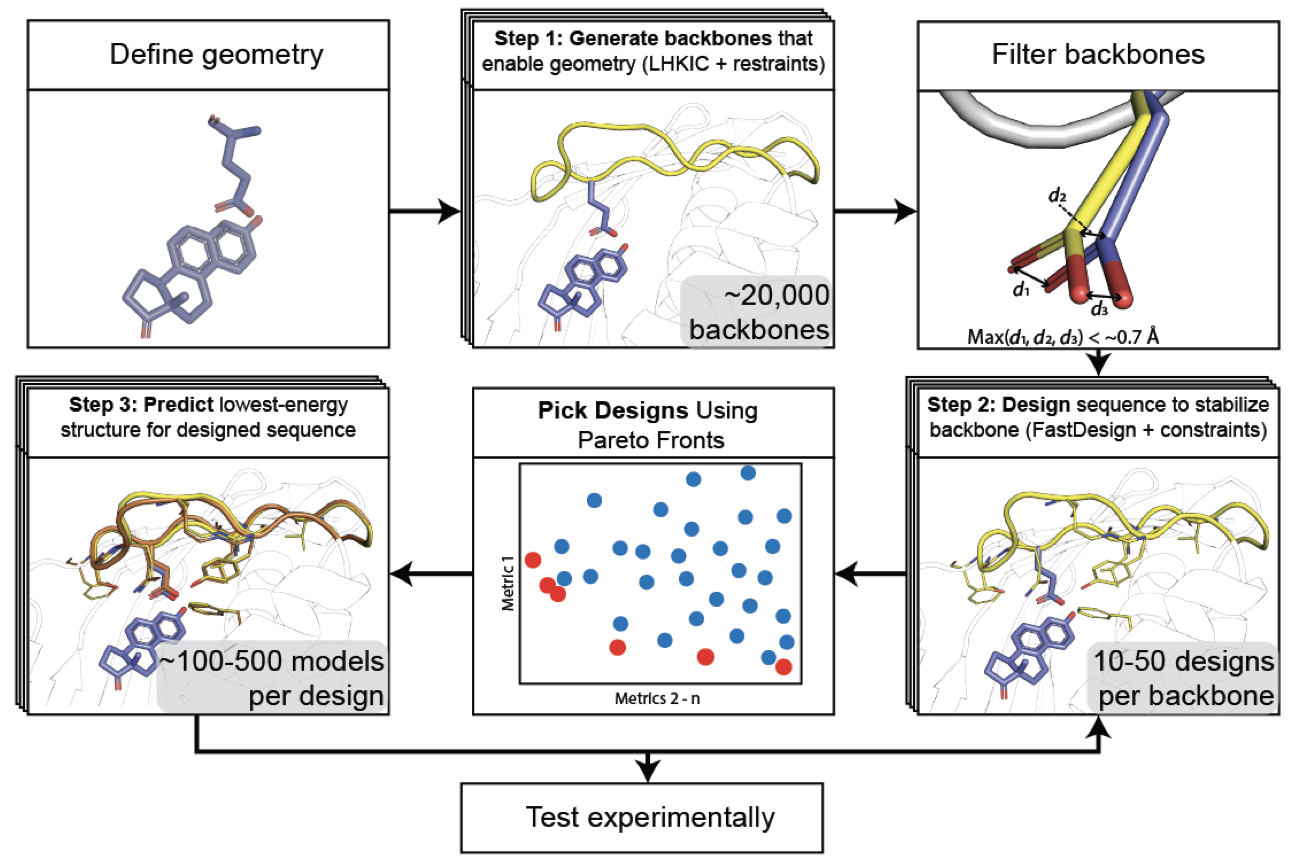
- PMID: 35254891
- PMCID: PMC8931229
- BioRxiv Preprint:
- Full Text
- Deposited Structures: 6UAD, 6UAE
- Gray lab @ Johns Hopkins University
- Thompson lab @ UC Merced
- Kortemme lab @ UC San Francisco
Access the paper
Additional Links
Structural basis for context-specific inhibition of translation by oxazolidinone antibiotics
Tsai K*, Stojković V*, Lee DJ*, Young ID, Szal T, Vazquez-Laslop N, Mankin AS, Fraser JS, Fujimori DG
Nature Structural and Molecular Biology, 2022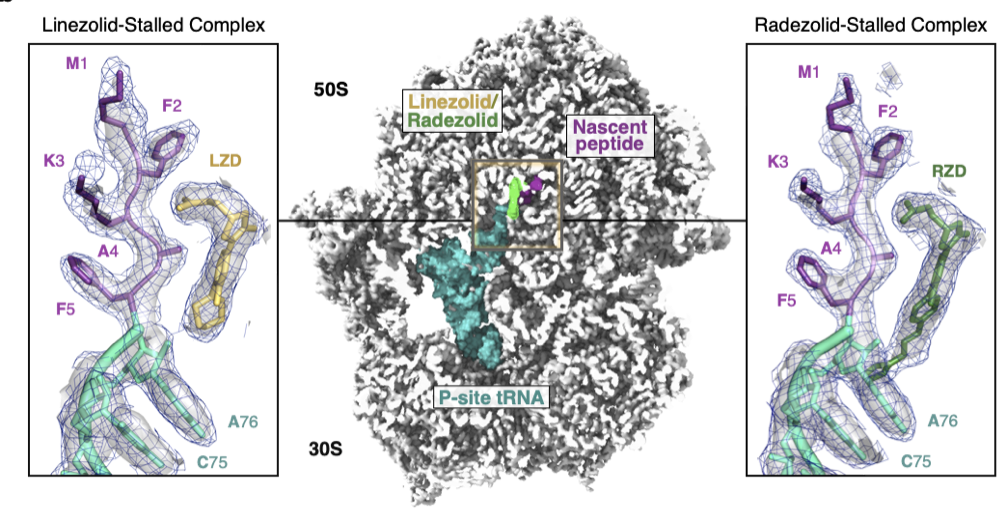
- PMID: 35165456
- PMCID: PMC8906282
- BioRxiv Preprint: 455846
- Full Text
- Deposited Structures and Maps: 7S1G/24800, 7S1H/24801, 7S1I/24802, 7S1J/24803, 7S1K/24804
- Fujimori lab @ UC San Francisco
- Mankin and Vazquez-Laslop lab @ University of Illinois at Chicago
- News and Views - Putting the antibiotics chloramphenicol and linezolid into context
Access the paper
Additional Links
Integration of software tools for integrative modeling of biomolecular systems
Hancock M, Peulen TO, Webb B, Poon B, Fraser JS, Adams P, Sali A
Journal of Structural Biology, 2022
- PMID: 35149213
- PMCID: PMC9278553
- BioRxiv Preprint:
- Full Text
- Adams lab @ Lawrence Berkeley National Laboratory
- Sali lab @ UC San Francisco
Access the paper
Additional Links
A counter-enzyme complex regulates glutamate metabolism in Bacillus subtilis
Jayaraman V, Lee DJ, Elad N, Vimer S, Sharon M, Fraser JS, Tawfik DS
Nature Chemical Biology, 2022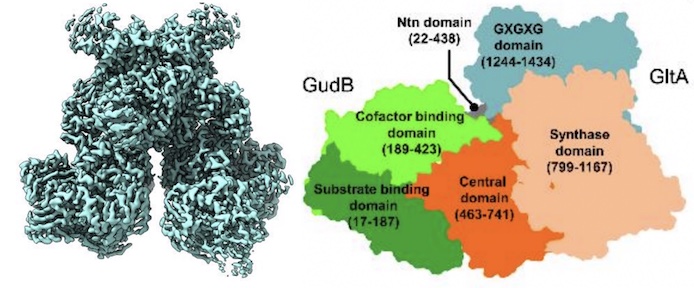
- PMID: 34931064
- PMCID: PMC8810680
- BioRxiv Preprint: 439528
- Full Text
- Deposited Structures and Maps: 7MFM/23817, 7MFT/23825
- Tawfik lab @ Weizmann Institute
- Dan Salah Tawfik (1955-2021)—A giant of protein evolution
- Celebratory Tweetstorm by first author Vijay Jayaraman
- News and Views - A complex struggle for direction
- News and Views - The Bacillus subtilis glutamate anti-metabolon
Access the paper
Additional Links
Directed evolution of the rRNA methylating enzyme Cfr reveals molecular basis of antibiotic resistance
Tsai K, Stojković V, Noda-Garcia L, Young ID, Myasnikov AG, Kleinman J, Palla A, Floor SN, Frost A, Fraser JS, Tawfik DS, Fujimori DG
eLife, 2022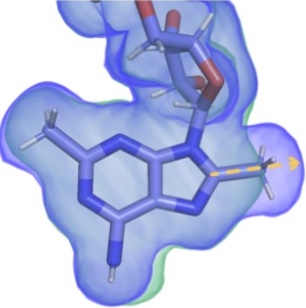
- PMID: 35015630
- PMCID: PMC8752094
- BioRxiv Preprint: 435202
- Full Text
- Deposited Structure and Map: 7LVK/23539
- GitHub Repository: fraser-lab/qptm (qPTxM)
- Tawfik lab @ Weizmann Institute
- Fujimori lab @ UC San Francisco
Access the paper
Additional Links
CryoEM and AI reveal a structure of SARS-CoV-2 Nsp2, a multifunctional protein involved in key host processes
Gupta M*, Azumaya CM*, Moritz M*, Pourmal S*, Diallo A*, Merz GE*, Jang G*, Bouhaddou M*, Fossati*, Brilot AF, Diwanji D, Hernandez E, Herrera N, Kratochvil HT, Lam VL, Li F, Li Y, Nguyen HC, Nowotny C, Owens TW, Peters JK, Rizo AN, Schulze-Gahmen U, Smith AM, Young ID, Yu Z, Asarnow D, Billesbølle C, Campbell MG, Chen J, Chen KH, Chio US, Dickinson MS, Doan L, Jin M,, Kim K, Li J, Li YL, Linossi E, Liu Y, Lo M, Lopez J, Lopez KE, Mancino A, Moss III FR, Paul MD, Pawar KI, Pelin A, Pospiech Jr. TH, Puchase C, Remesh SG, Safari M, Schaefer K, Sun M, Tabios MC, Thwin AC, Titus EW, Trenker R, Tse E, Tsui TKM, Wang F, Zhang K, Zhang Y, Zhao J, Zhou F, Zhou Y, Zuliani-Alvarez L, QCRG Structural Biology Consortium, Agard DA, Cheng Y, Fraser JS, Jura N, Kortemme T, Manglik A, Southworth DR, Stroud RM, Swaney DL, Krogan NJ, Frost A, Rosenberg OS, Verba KA
Biorxiv, 2021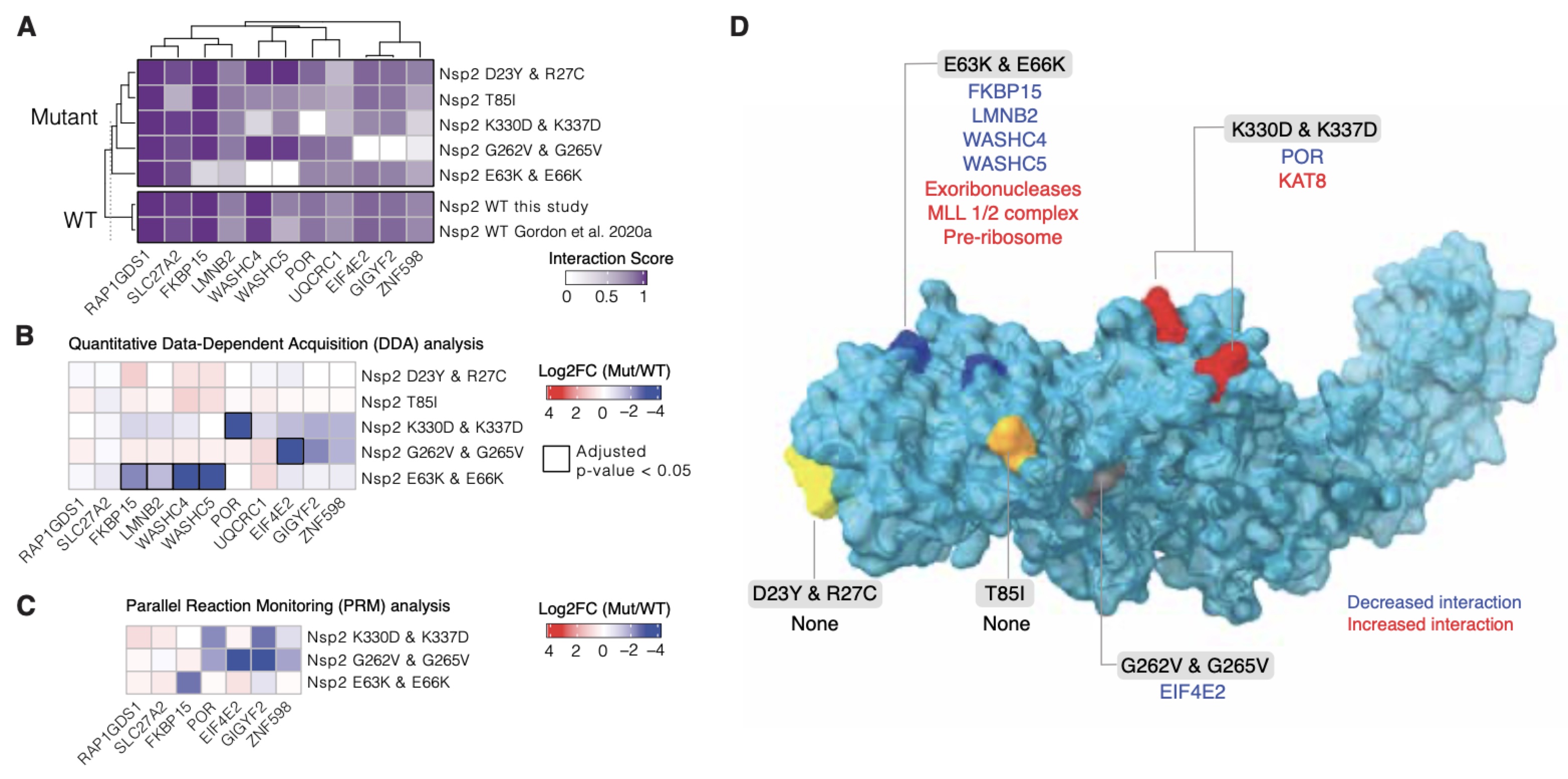
- BioRxiv Preprint: 443524
- Deposited Structures: 7MSW, 7MSX
- QBI Coronavirus Research Group @ UC San Francisco
Access the paper
Additional Link
State of the structure address on MET receptor activation by HGF
Linossi EM, Estevam GO, Oshima M, Fraser JS, Collisson EA, Jura N
Biochemical Society Transactions, 2021
- PMID: 33860789
- PMCID: PMC8711257
- Full Text
- Jura lab @ UC San Francisco
- Collisson lab @ UC San Francisco
Access the paper
Additional Links
Fragment binding to the Nsp3 macrodomain of SARS-CoV-2 identified through crystallographic screening and computational docking
Schuller M*, Correy GJ*, Gahbauer S*, Fearon D*, Wu T, Díaz RE, Young ID, Carvalho Martins L, Smith DS, Schulze-Gahmen U, Owens TW, Deshpande I, Merz GE, Thwin AC, Biel JT, Peters JK, Mortiz M, Herrera N, Kratochvil HT, QCRG Structural Biology Consortium, Aimon A, Bennett MJ, Brandao Neto J, Cohen EA, Dias A, Douangamath A, Dunnett L, Fedorov O, Ferla PM, Fuchs M, Gorrie-Stone JT, Holton MJ, Johnson GM, Krojer T, Meigs G, Powell JA, Rack J, Rangel LV, Russi S, Skyner ER, Smith AC, Soares SA, Wierman LJ, Zhu K, O’Brien P, Jura N, Ashworth A, Irwin J, Thompson MC, Gestwicki JE, von Delft F, Shoichet BK, Fraser JS, Ahel I
Science Advances, 2021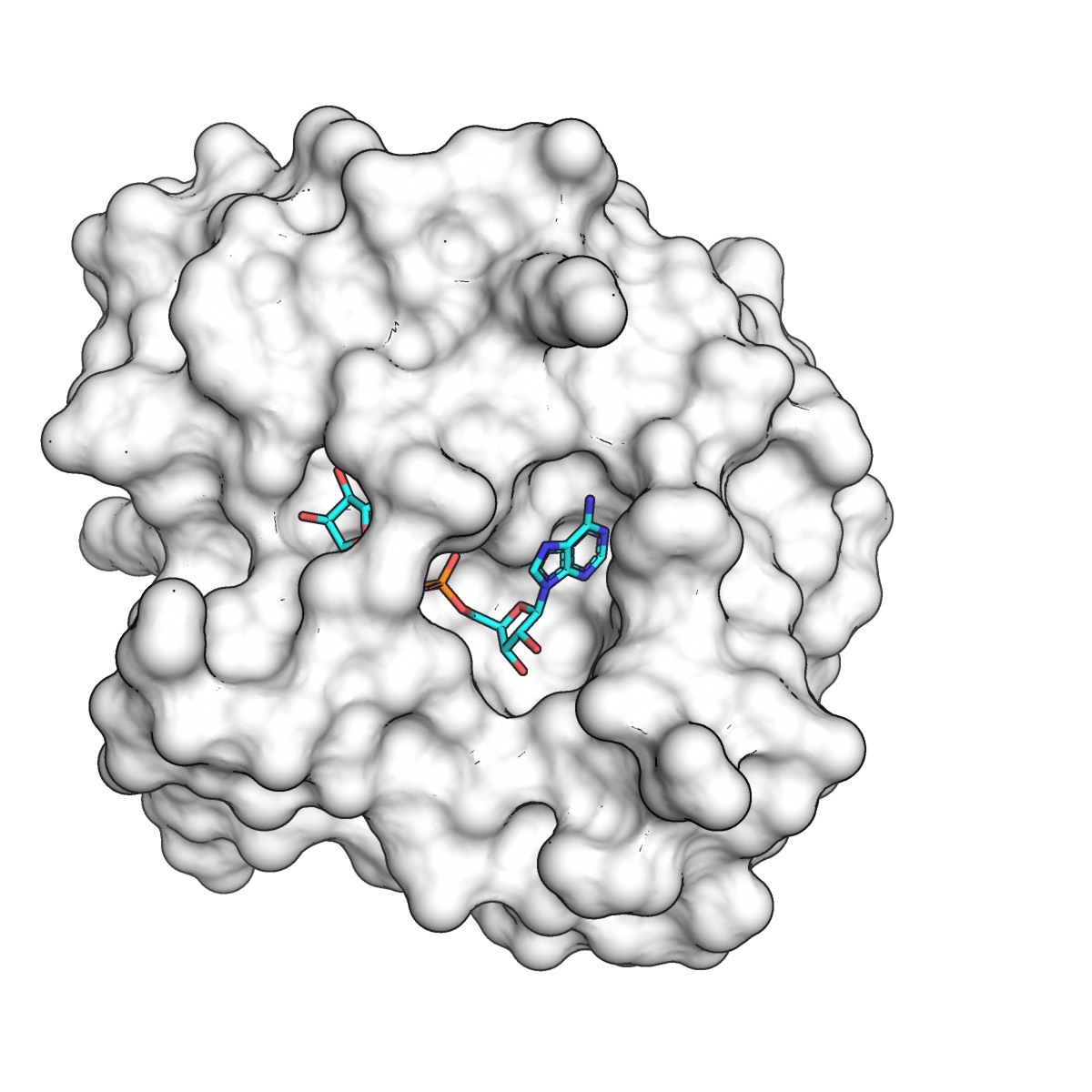
- PMID: 33853786
- PMCID: PMC8046379
- BioRxiv Preprint: 393405
- Full Text
- Deposited Structures: 7KR0, 7KR1, 7KQW, 7KQO, 7KQP, 5RVJ, 5RVK, 5RVL, 5RVM, 5RVN, 5RVO, 5RVP, 5RVQ, 5RVR, 5RVS, 5RVT, 5RVU, 5RVV, 5RS7, 5RS8, 5RS9, 5RSB, 5RSC, 5RSD, 5RSE, 5RSF, 5RSG, 5RSH, 5RSI, 5RSJ, 5RSK, 5RSL, 5RSM, 5RSN, 5RSO, 5RSP, 5RSQ, 5RSR, 5RSS, 5RST, 5RSU, 5RSV, 5RSW, 5RSX, 5RSY, 5RSZ, 5RT0, 5RT1, 5RT2, 5RT3, 5RT4, 5RT5, 5RT6, 5RT7, 5RT8, 5RT9, 5RTA, 5RTB, 5RTC, 5RTD, 5RTE, 5RTF, 5RTG, 5RTH, 5RTI, 5RTJ, 5RTK, 5RTL, 5RTM, 5RTN, 5RTO, 5RTP, 5RTQ, 5RTR, 5RTS, 5RTT, 5RTU, 5RTV, 5RTW, 5RTX, 5RTY, 5RTZ, 5RU0, 5RU1, 5RU2, 5RU3, 5RU4, 5RU5, 5RU6, 5RU7, 5RU8, 5RU9, 5RUA, 5RUC, 5RUD, 5RUE, 5RUF, 5RUG, 5RUH, 5RUI, 5RUJ, 5RUK, 5RUL, 5RUM, 5RUN, 5RUO, 5RUP, 5RUQ, 5RUR, 5RUS, 5RUT, 5RUU, 5RUV, 5RUW, 5RUX, 5RUY, 5RUZ, 5RV0, 5RV1, 5RV2, 5RV3, 5RV4, 5RV5, 5RV6, 5RV7, 5RV8, 5RV9, 5RVA, 5RVB, 5RVC, 5RVD, 5RVE, 5RVF, 5RVG, 5RVH, 5RVI, 5S6W, 5S18, 5S1A, 5S1C, 5S1E, 5S1G, 5S1I, 5S1K, 5S1M, 5S1O, 5S1Q, 5S1S, 5S1U, 5S1W, 5S1Y, 5S20, 5S22, 5S24, 5S26, 5S27, 5S28, 5S29, 5S2A, 5S2B, 5S2C, 5S2D, 5S2E, 5S2F, 5S2G, 5S2H, 5S2I, 5S2J, 5S2K, 5S2L, 5S2M, 5S2N, 5S2O, 5S2P, 5S2Q, 5S2R, 5S2S, 5S2T, 5S2U, 5S2V, 5S2W, 5S2X, 5S2Y, 5S2Z, 5S30, 5S31, 5S32, 5S33, 5S34, 5S35, 5S36, 5S37, 5S38, 5S39, 5S3A, 5S3B, 5S3C, 5S3D, 5S3E, 5S3F, 5S3G, 5S3H, 5S3I, 5S3J, 5S3K, 5S3L, 5S3M, 5S3N, 5S3O, 5S3P, 5S3Q, 5S3R, 5S3S, 5S3T, 5S3U, 5S3V, 5S3W, 5S3X, 5S3Y, 5S3Z, 5S40, 5S41, 5S42, 5S43, 5S44, 5S45, 5S46, 5S47, 5S48, 5S49, 5S4A, 5S4B, 5S4C, 5S4D, 5S4E, 5S4F, 5S4G, 5S4H, 5S4I, 5S4J, 5S4K
- Zenodo Records: 4716363 (UCSF C2 crystal form screen), 4716089 (UCSF P43 crystal form screen), 4606901 (Docking Fragment Screen), 4628855 (Differential Scanning Fluorimetry), 4628875 (Isothermal Titration Calorimetry)
- Addgene Plasmids: 169209 (C2 crystal form), 169210 (P43 crystal form)
- Original post
- Fraganalysis from Diamond Lightsource
- QBI Coronavirus Research Group @ UC San Francisco
- von Delft lab @ University of Oxford
- Ahel lab @ University of Oxford
- Celebratory Tweetstorm by James Fraser
- Celebratory Tweetstorm by Mike Thompson
- UCSF Press Release - Building Blocks for COVID-19 Antiviral Drugs Identified in Rapid Study
- Practical Fragments - Hundreds of fragments hits for the SARS-CoV-2 Nsp3 Macrodomain
- Massive fragment screen points way to new SARS-CoV-2 inhibitors
Access the paper
Additional Links
Cryo-EM model validation recommendations based on outcomes of the 2019 EMDataResource challenge
Lawson CL, Kryshtafovych A, Adams PD, Afonine PV, Baker ML, Barad BA, Bond P, Burnley T, Cao R, Cheng J, Chojnowski G, Cowtan K, Dill KA, DiMaio F, Farrell DP, Fraser JS, Jr. Herzik MA, Hoh SW, Hou J, Hung L, Igaev M, Joseph AP, Kihara D, Kumar D, Mittal S, Monastyrskyy B, Olek M, Palmer CM, Patwardhan A, Perez A, Pfab J, Pintilie GD, Richardson JS, Rosenthal PB, Sarkar D, Schäfer LU, Schmid MF, Schröder GF, Shekhar M, Si D, Singharoy A, Terashi G, Terwilliger TC, Vaiana A, Wang L, Wang Z, Wankowicz SA, Williams CJ, Winn M, Wu T, Yu X, Zhang K, Berman HM, Chiu W
Nature Methods, 2021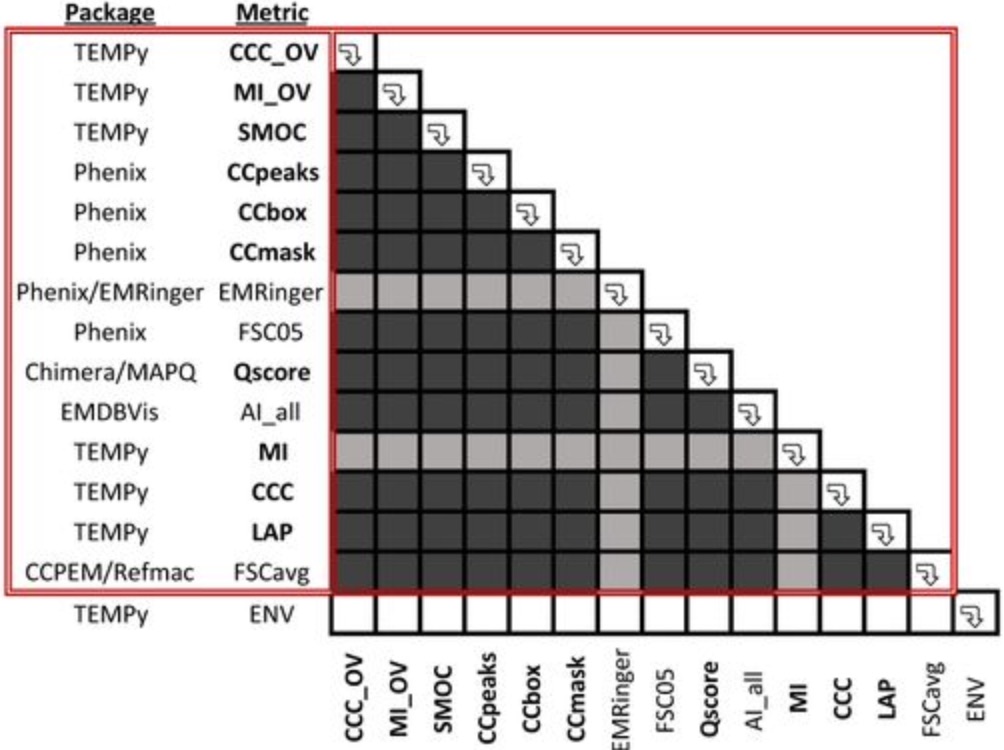
- PMID: 33542514
- PMCID: PMC7864804
- BioRxiv Preprint: 147033
- Full Text
- Zenodo Record: 4148789 (2019 EMDataResource Model Challenge Dataset)
- 2019 Model Metrics Challenge
- News and Views - Improving cryo-EM structure validation
Access the paper
Additional Links
qFit 3: Protein and ligand multiconformer modeling for X-ray crystallographic and single-particle cryo-EM density maps
Riley BT, Wankowicz SA, de Oliveira SHP, van Zundert GCP, Hogan DW, Fraser JS, Keedy DA, van den Bedem H
Protein Science, 2021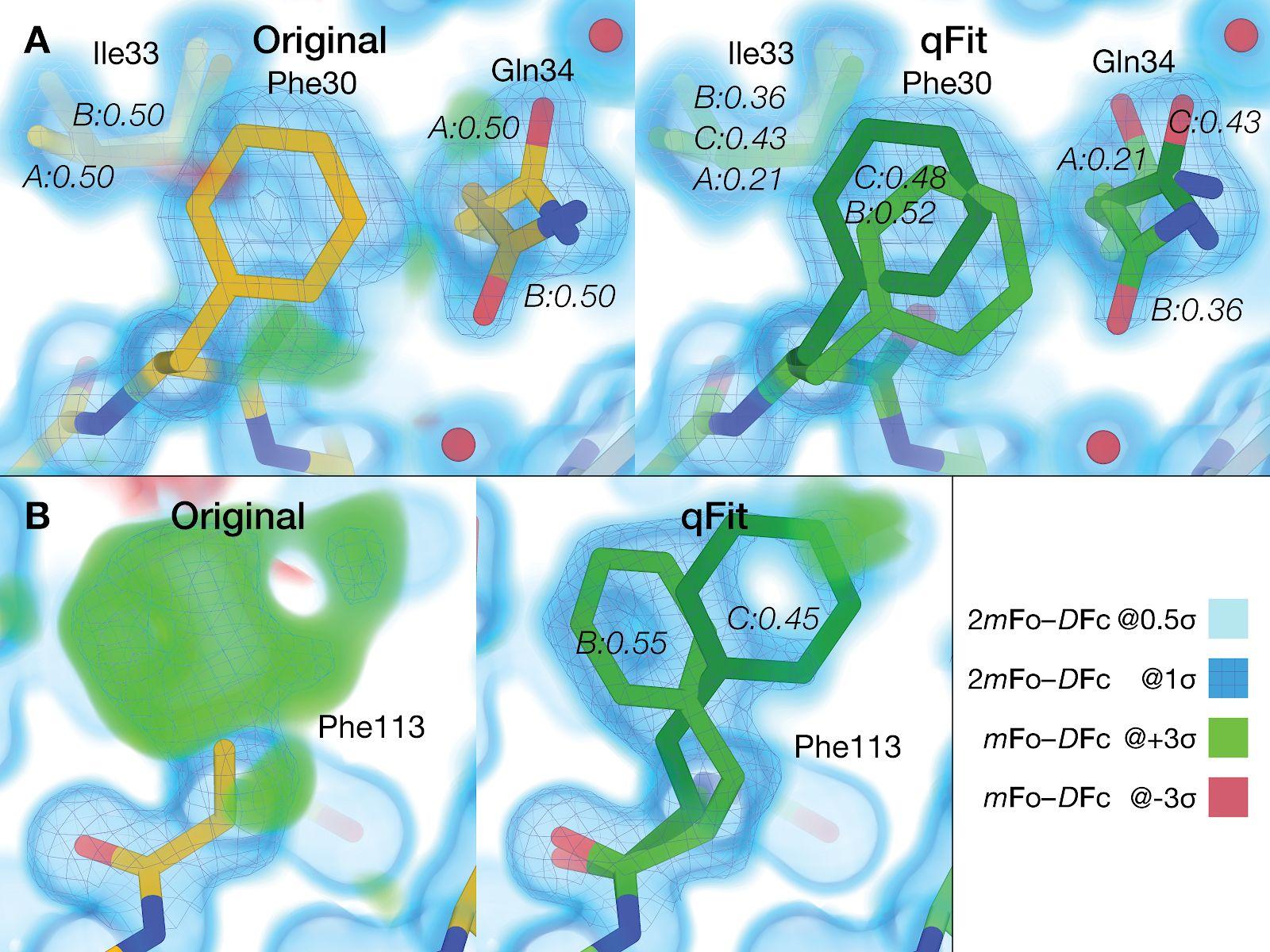
- PMID: 33210433
- PMCID: PMC7737783
- BioRxiv Preprint: 280222
- Full Text
- Zenodo Record: 4021213 (qfit-3.0: v3.2.0)
- GitHub Repository: ExcitedStates/qfit-3.0
- Keedy lab @ CUNY Advanced Science Research Center
Access the paper
Additional Link
Assessment of enzyme active site positioning and tests of catalytic mechanisms through X-ray–derived conformational ensembles
Yabukarski F, Biel JT, Pinney MM, Doukov T, Powers AS, Fraser JS, Herschlag D
PNAS, 2020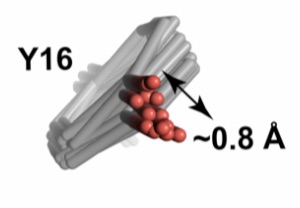
Genetic interaction mapping informs integrative structure determination of protein complexes
Braberg H*, Echeverria I*, Bohn S*, Cimermancic P*, Shiver A, Alexander R, Xu J, Shales M, Dronamraju R, Jiang S, Dwivedi G, Bogdanoff D, Chaung KK, Hüttenhain R, Wang S, Mavor D, Pellarin R, Schneidman D, Bader JS, Fraser JS, Morris J, Haber JE, Strahl BD, Gross CA, Dai J, Boeke JD, Sali A, Krogan NJ
Science, 2020
Discovery of allosteric binding sites by crystallographic fragment screening
Krojer T, Fraser JS, von Delft F
Current Opinions in Structural Biology, 2020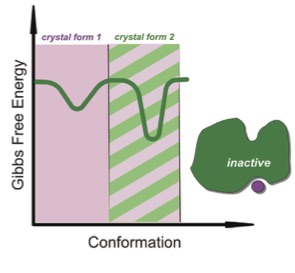
- PMID: 33171388
- PMCID: PMC10979522
- Full Text
- XChem - Fragment Screening @ Diamond Light Source
Access the paper
Additional Link
Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms
Gordon DE*, Hiatt J*, Bouhaddou M*, Rezelj VV*, Ulferts S*, Braberg H*, Jureka AS*, Obernier K*, Guo JZ*, Batra J*, Kaake RM*, Weckstein AR*, Owens TW*, Gupta M*, Pourmal S*, Titus EW*, Cakir M*, Soucheray M, McGregor M, Cakir Z, Jang G, O’Meara MJ, Tummino TA, Zhang Z, Foussard H, Rojc A, Zhou Y, Kuchenov D, Hüttenhain R, Xu J, Eckhardt M, Swaney DL, Fabius JM, Ummadi M, Tutuncuoglu B, Rathore U, Modak M, Haas P, Haas KM, Naing ZZC, Pulido EH, Shi Y, Barrio-Hernandez I, Memon D, Petsalaki E, Dunham A, Marrero MC, Burke D, Koh C, Vallet T, Silvas JA, Azumaya CM, Billesbølle C, Brilot AF, Campbell MG, Diallo A, Dickinson MS, Diwanji D, Herrera N, Hoppe N, Kratochvil HT, Liu Y, Merz GE, Moritz M, Nguyen HC, Nowotny C, Puchades C, Rizo AN, Schulze-Gahmen U, Smith AM, Sun M, Young ID, Zhao J, Asarnow D, Biel J, Bowen A, Braxton JR, Chen J, Chio CM, Chio US, Deshpande I, Doan L, Faust B, Flores S, Jin M, Kim K, Lam VL, Li F, Li J, Li YL, Li Y, Liu X, Lo M, Lopez KE, Melo AA, Moss FR 3rd, Nguyen P, Paulino J, Pawar KI, Peters JK, Pospiech TH Jr, Safari M, Sangwan S, Schaefer K, Thomas PV, Thwin AC, Trenker R, Tse E, Tsui TKM, Wang F, Whitis N, Yu Z, Zhang K, Zhang Y, Zhou F, Saltzberg D, QCRG Structural Biology Consortium, Hodder AJ, Shun-Shion AS, Williams DM, White KM, Rosales R, Kehrer T, Miorin L, Moreno E, Patel AH, Rihn S, Khalid MM, Vallejo-Gracia A, Fozouni P, Simoneau CR, Roth TL, Wu D, Karim MA, Ghoussaini M, Dunham I, Berardi F, Weigang S, Chazal M, Park J, Logue J, McGrath M, Weston S, Haupt R, Hastie CJ, Elliott M, Brown F, Burness KA, Reid E, Dorward M, Johnson C, Wilkinson SG, Geyer A, Giesel DM, Baillie C, Raggett S, Leech H, Toth R, Goodman N, Keough KC, Lind AL; Zoonomia Consortium, Klesh RJ, Hemphill KR, Carlson-Stevermer J, Oki J, Holden K, Maures T, Pollard KS, Sali A, Agard DA, Cheng Y, Fraser JS, Frost A, Jura N, Kortemme T, Manglik A, Southworth DR, Stroud RM, Alessi DR, Davies P, Frieman MB, Ideker T, Abate C, Jouvenet N, Kochs G, Shoichet B, Ott M, Palmarini M, Shokat KM, García-Sastre A, Rassen JA, Grosse R, Rosenberg OS, Verba KA, Basler CF, Vignuzzi M, Peden AA, Beltrao P, Krogan NJ
Science, 2020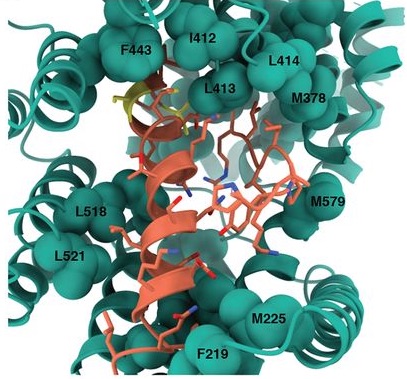
- PMID: 33060197
- PMCID: PMC7808408
- Full Text
- Deposited Structure and Map: 7KDT/22829
- NIH Director's Blog - Protein Mapping Study Reveals Valuable Clues for COVID-19 Drug Development
- QBI Coronavirus Research Group @ UC San Francisco
Access the paper
Additional Links
Synthetic group A streptogramin antibiotics that overcome Vat resistance
Li Q*, Pellegrino J*, Lee DJ, Tran AA, Chaires HC, Wang R, Park JE, Ji K, Chow D, Zhang N, Brilot AF, Biel JT, van Zundert G, Borrelli K, Shinabarger D, Wolfe C, Murray B, Jacobson MP, Mühle E, Chesneau O, Fraser JS, Seiple IB
Nature, 2020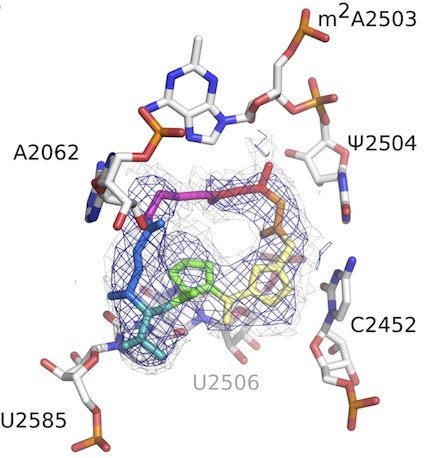
- PMID: 32968273
- PMCID: PMC7546582
- ChemRxiv Preprint: 8346107
- Full Text
- Deposited Structures: 6X3C, 6X3J
- Deposited Structures, Maps, and Data sets: 6PCH/20300/10522, 6PCQ/20304/10521, 6PCR/20305/10524, 6PCS/20306/10523, 6PCT/20307/10526, 6PC5/20296/10530, 6PC6/20297/10529, 6PC7/20298/10527, 6PC8/20299/10525, 6WYV/21969/10528
- News and Views - Modular synthesis enables molecular ju-jitsu in the fight against antibiotic resistance
- C&EN - How to outwit antibiotic resistance with chemical know-how
- UCSF Press Release - Customizable Synthetic Antibiotic Outmaneuvers Resistant Bacteria
- Seiple lab @ UC San Francisco
- Schrodinger Inc.
- Paper Submission Celebration Photo
Access the paper
Additional Links
Ensemble-based enzyme design can recapitulate the effects of laboratory directed evolution in silico
Broom A*, Rakotoharisoa RV*, Thompson MC, Zarifi N, Nguyen E, Mukhametzhanov N, Liu L, Fraser JS, Chica RA
Nature Communications, 2020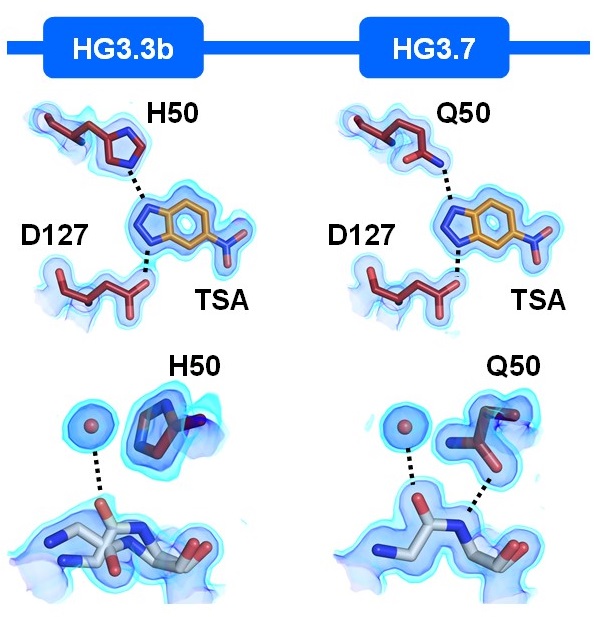
- PMID: 32968058
- PMCID: PMC7511930
- BioRxiv Preprint: 999235
- Full Text
- Deposited Structures: 5RG4, 5RG5, 5RG7, 5RG6, 5RG8, 5RG9, 5RGA, 5RGB, 5RGC, 5RGD, 5RGE, 5RGF
- Chica lab @ University of Ottawa
- Faculty Opinions
- Rise of the mutants - New uOttawa-led research to improve enzyme design methodologies
- Nature Catalysis - Ensemble-based enzyme design
Access the paper
Additional Links
Expanding the space of protein geometries by computational design of de novo fold families
Pan X, Thompson MC, Zhang Y, Liu L, Fraser JS, Kelly MJS, Kortemme T
Science, 2020
- PMID: 32855341
- PMCID: PMC7787817
- BioRxiv Preprint: 041772
- Full Text
- Deposited Structures: 6VG7, 6VGA, 6VGB, 6W90
- Kortemme lab @ UC San Francisco
Access the paper
Additional Link
A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing
Gordon DE*, Jang GM*, Bouhaddou M*, Xu J*, Obernier K*, White KM*, O’Meara MJ*, Rezelj VV*, Guo JZ, Swaney DL, Tummino TA, Huttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Lyu J, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Rakesh R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Liboy-Lugo J, Lin Y, Huang XP, Liu Y, Wankowicz SA, Bohn M, Safari M, Ugur FS, Koh C, Savar NS, Tran QD, Shengjuler D, Fletcher SJ, O’Neal MC, Cai Y, Chang JCJ, Broadhurst DJ, Klippsten S, Sharp PP, Wenzell NA, Kuzuoglu D, Wang HY, Trenker R, Young JM, Cavero DA, Hiatt J, Roth TL, Rathore U, Subramanian A, Noack J, Hubert M, Stroud RM, Frankel AD, Rosenberg OS, Verba KA, Agard D, Ott M, Emerman M, Jura N, von Zastrow M, Verdin E, Ashworth A, Schwartz O, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor S, Fraser JS, Gross JD, Sali A, Roth BL, Ruggero D, Taunton J, Kortemme T, Beltrao P, Vignuzzi M, García-Sastre A, Shokat KM, Shoichet BK, Krogan NJ
Nature, 2020
- PMID: 32353859
- PMCID: PMC7431030
- BioRxiv Preprint: 002386
- Full Text
- GitHub Repository: fraser-lab/QCRG_COVID19_Figures (gtex/gnomAD figure scripts)
- QBI Coronavirus Research Group @ UC San Francisco
- Celebratory Tweetstorm by James Fraser
- New York Times: QBI Coronavirus Research Group
- Science Magazine: What the Coronavirus Proteins Are Targeting
- UCSF: Unveiling How COVID-19 Hijacks Our Cells to Help Rush New Drugs to Patients
- New York Times: Old Drugs May Find a New Purpose: Fighting the Coronavirus.
Access the paper
Additional Links
Assessment of the nucleotide modifications in the high-resolution cryo-electron microscopy structure of the Escherichia coli 50S subunit
Stojković V, Myasnikov AG, Young ID, Frost A, Fraser JS, Fujimori DG
Nucleic Acids Research, 2020
- PMID: 31989172
- PMCID: PMC7049716
- BioRxiv Preprint: 695429
- Full Text
- Deposited Structure and Map: 6PJ6/20353
- Zenodo Record: 3555658 (QPTxM code at time of publication)
- GitHub Repository: fraser-lab/qptm (qPTxM)
- Frost lab @ UC San Francisco
- Fujimori lab @ UC San Francisco
Access the paper
Additional Links
Comparing serial X-ray crystallography and microcrystal electron diffraction (MicroED) as methods for routine structure determination from small macromolecular crystals
Wolff AM, Young ID, Sierra RG, Brewster AS, Martynowycz MW, Nango E, Sugahara M, Nakane T, Ito K, Aquila A, Bhowmick A, Biel JT, Carbajo S, Cohen AE, Cortez S, Gonzalez A, Hino T, Im D, Koralek JD, Kubo M, Lazarou TS, Nomura T, Owada S, Samelson A, Tanaka R, Tanaka T, Thompson EM, van den Bedem H, Woldeyes RA, Yumoto F, Zhao W, Tono K, Boutet S, Iwata S, Gonen T, Sauter NK, Fraser JS, Thompson MC
IUCrJ, 2020
- PMID: 32148858
- PMCID: PMC7055375
- BioRxiv Preprint: 767061
- Full Text
- Online Datasets:
- Deposited Structures: 6U5C, 6U5D, 6U5E
- Deposited Structure and Map: 6U5G/20645
- Gonen lab @ UC Los Angeles
- LCLS MFX Beamline
- SACLA XFEL
- Celebratory Tweetstorm by first author Alex Wolff
- Celebratory Tweetstorm by corresponding author Michael Thompson
Access the paper
Additional Links
What will computational modelling approaches have to say in the era of atomistic cryo-EM data?
Fraser JS, Lindorff-Larsen K, Bonomi M
Journal of Chemical Information and Modeling, 2020
- PMID: 32090567
- PMCID: PMC8561786
- arXiv Preprint: 2002.00151
- Full Text
- Kresten Lindorff-Larsen @ University of Copenhagen
- Max Bonomi @ The Institut Pasteur
Access the paper
Additional Links
Co-occurring alterations in the RAS-MAPK pathway limit response to MET inhibitor treatment in MET exon 14 skipping mutation-positive lung cancer
Rotow JK, Gui P, Wu W, Raymond VM, Lanman RB, Kaye FJ, Peled N, Fece de la Cruz F, Nadres B, Corcoran RB, Yeh I, Bastian BC, Starostik P, Newsom K, Olivas V, Wolff AM, Fraser JS, Collisson EA, McCoach CE, Camidge DR, Pacheco J, Bazhenova L, Li T, Bivona TG, Blakely CM
Clinical Cancer Research, 2020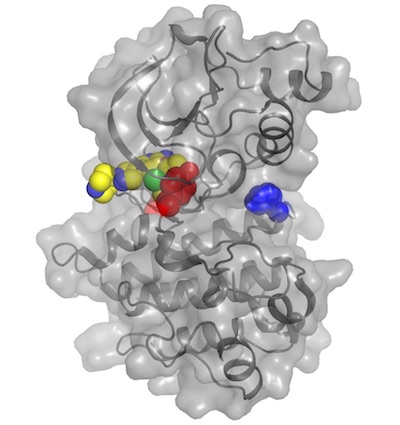
- PMID: 31548343
- PMCID: PMC6980768
- BioRxiv Preprint: 374181
- Full Text
- Bivona lab @ UC San Francisco
Access the paper
Additional Link
Differences in the chitinolytic activity of mammalian chitinases on soluble and insoluble substrates
Barad BA, Liu L, Diaz RE, Basillo R, Van Dyken SJ, Locksley RM, Fraser JS
Protein Science, 2020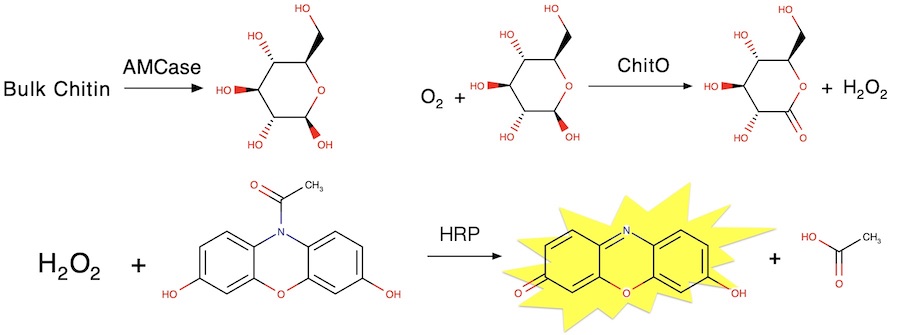
- PMID: 31930591
- PMCID: PMC7096708
- BioRxiv Preprint: 762336
- Full Text
- GitHub Repositories: fraser-lab/chitin_analysis (Kinetic Analysis of Chitinases), fraser-lab/relax (Calculating non-linear relaxation fits)
- Locksley lab @ UC San Francisco
- Van Dyken lab @ Washington University in St. Louis
- Celebratory Tweetstorm by first author Ben Barad
Access the paper
Additional Links
Ensemble refinement produces consistent R-free values but smaller ensemble sizes than previously reported
Wankowicz SA, Fraser JS
Computational Crystallography Newsletter, 2020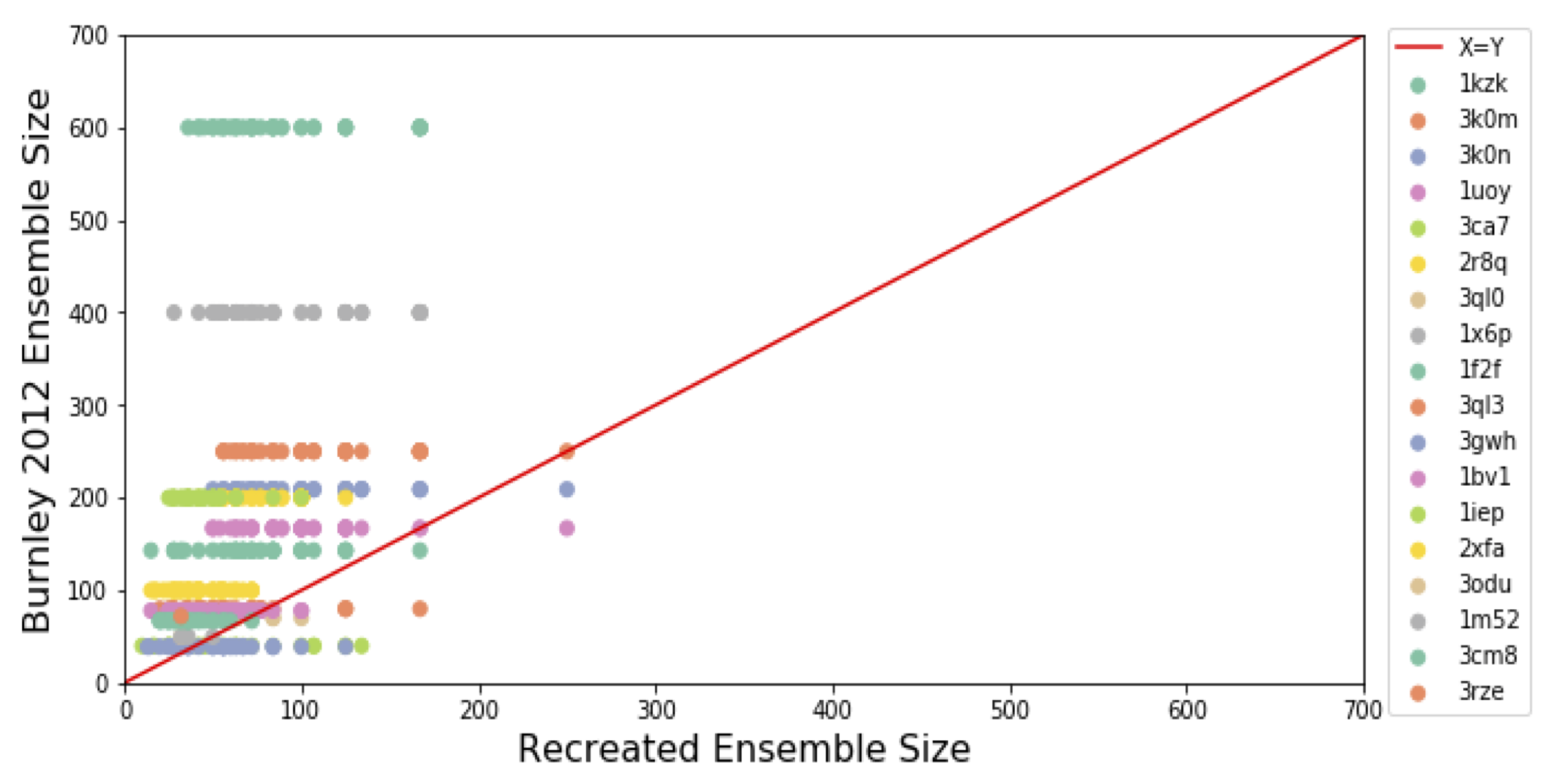
- Full Text
- PHENIX
- PHENIX Computational Crystallography Newsletter
Access the paper
Additional Links
Liquid-like and rigid-body motions in molecular dynamics simulations of a crystalline protein
Wych DC, Fraser JS, Mobley DL, Wall ME
Structural Dynamics, 2019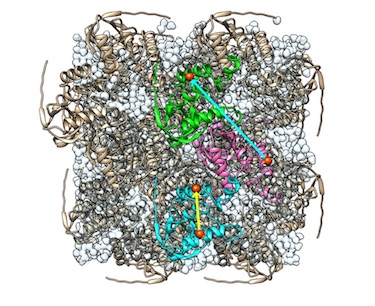
- PMID: 31867408
- PMCID: PMC6920053
- BioRxiv Preprint: 811083
- Full Text
- Mobley lab @ UC Irvine
- Michael Wall @ Los Alamos National Lab
- Scilight - Molecular dynamics approach observes rigid-body and liquid-like motion in protein crystals
Access the paper
Additional Links
Mix-and-inject XFEL crystallography reveals gated conformational dynamics during enzyme catalysis
Dasgupta M, Budday D, Oliveira SHP, Madzelan P, Marchany-Rivera D, Seravalli J, Hayes B, Sierra RG, Boutet S, Hunter MS, Alonso-Mori R, Batyuk A, Wierman J, Lyubimov A, Brewster AS, Sauter NK, Applegate GA, Tiwari VK, Berkowitz DB, Thompson MC, Cohen AE, Fraser JS, Wall ME, van den Bedem H, Wilson MA
PNAS, 2019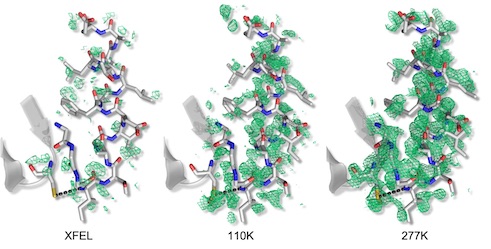
- PMID: 31801874
- PMCID: PMC6926069
- BioRxiv Preprint: 524751
- Full Text
- Deposited Structures: 6NI4, 6NI9, 6NPQ, 6UND, 6UNF
- Henry van den Bedem @ Stanford University
- Wilson lab @ University of Nebraska-Lincoln
- Paper Submission Celebration Tweets
- SLAC: Researchers reveal how enzyme motions catalyze reactions
- University of Nebraska-Lincoln: Enzymes and X-rays - Study reveals hidden acrobatics of cellular catalysts
Access the paper
Additional Links
Computational design of a modular protein sense/response system
Glasgow AA*, Huang Y*, Mandell DJ*, Thompson M, Ritterson R, Loshbaugh AL, Pellegrino J, Krivacic C, Pache RA, Barlow KA, Ollikainen N, Jeon D, Kelly MJS, Fraser JS, Kortemme T
Science, 2019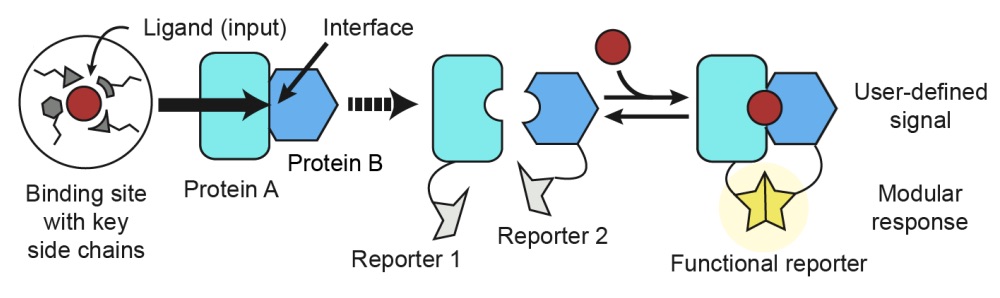
- PMID: 31754004
- PMCID: PMC7343396
- BioRxiv Preprint: 648485
- Full Text
- Deposited Structure: 6OB5
- Kortemme lab @ UC San Francisco
- SBGrid: Computational Design of Protein Sensors Coupled with Functional Outputs
- Science - Perspective: Designer sense-response systems
Access the paper
Additional Links
Temperature-Jump Solution X-ray Scattering Reveals Distinct Motions in a Dynamic Enzyme
Thompson MC, Barad BA, Wolff AM, Cho HS, Schotte F, Schwarz DMC, Anfinrud P, Fraser JS
Nature Chemistry, 2019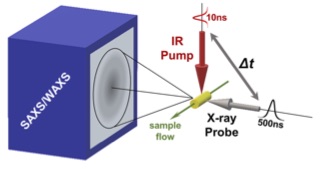
- PMID: 31527847
- PMCID: PMC6815256
- BioRxiv Preprint: 476432
- Full Text
- GitHub Repository: fraser-lab/solution_scattering (Solution Scattering)
- Solution Scattering Data on NIH Figshare
- Philip Anfinrud @ NIH
- BioCARS @ University of Chicago
- Paper Submission Celebration Tweetstorm
Access the paper
Additional Links
Synthetic essentiality of metabolic regulator PDHK1 in PTEN-deficient cells and cancers
Chatterjee N*, Pazarentzos E*, Mayekar MK, Gui P, Allegakoen DV, Hrustanovic G, Olivas V, Lin L, Verschueren E, Johnson JR, Hofree M, Yan JJ, Newton BW, Dollen JV, Earnshaw CH, Flanagan J, Chan E, Asthana S, Ideker T, Wu W, Suzuki J, Barad BA, Kirichok Y, Fraser JS, Weiss WA, Krogan NJ, Tulpule A, Sabnis AJ, Bivona TG
Cell Reports, 2019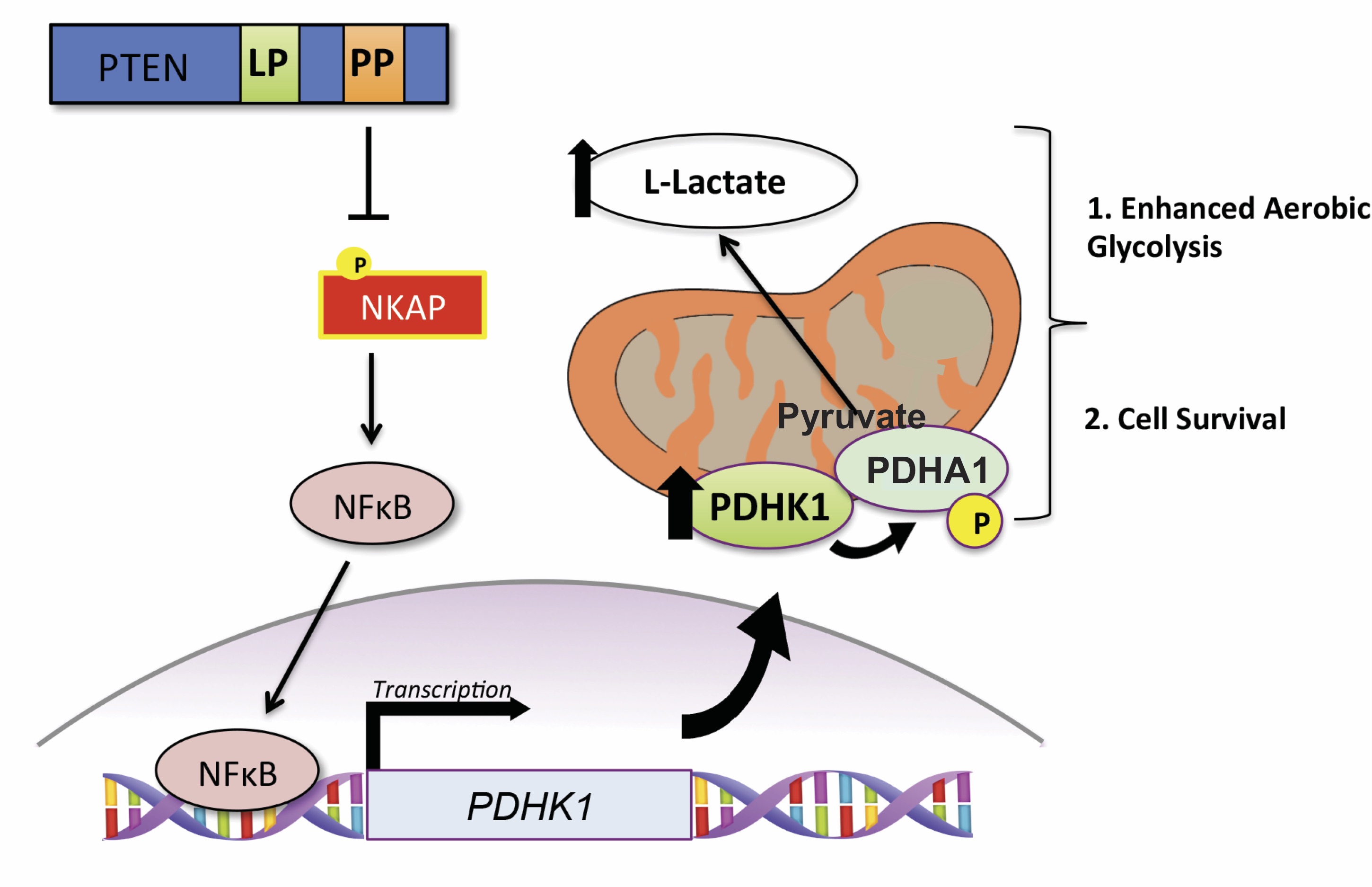
- PMID: 31461649
- PMCID: PMC6728083
- BioRxiv Preprint: 441295
- Full Text
- Bivona lab @ UC San Francisco
Access the paper
Additional Link
Effects of α-tubulin acetylation on microtubule structure and stability
Eshun-Wilson L, Zhang R, Portran D, Toso D, Lohr T, Vendruscolo M, Bonomi M, Fraser JS, Nogales E
PNAS, 2019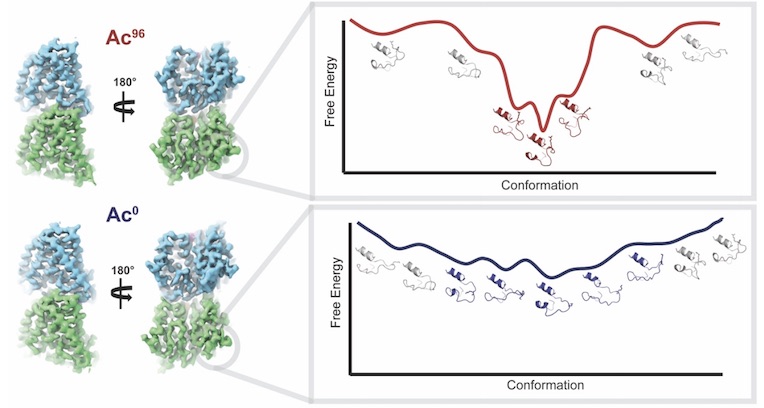
- PMID: 31072936
- PMCID: PMC6535015
- BioRxiv Preprint: 516591
- Full Text
- Deposited Structures and Maps: 6O2Q/0612, 6O2R/0613, 6O2S/0614, 6O2T/0615
- GitHub Repository: fraser-lab/plumed_em_md (PLUMED-EM)
- PLUMED-NEST files
- Nogales lab @ UC Berkeley
- Paper Submission Celebration Tweets
- SBGrid: Structure and Stability
Access the paper
Additional Links
A multi-model approach to assessing local and global cryo-EM map quality
Herzik Jr. MA, Fraser JS, Lander GC
Structure, 2019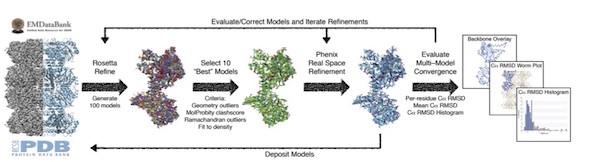
- PMID: 30449687
- PMCID: PMC6365196
- BioRxiv Preprint: 128561
- Full Text
- Convergence Server @ Lander lab
- F1000 evaluation
Access the paper
Additional Links
Biophysical characterization of a disabled double mutant of Soybean Lipoxygenase: the undoing of precise substrate positioning relative to metal cofactor and an identified dynamical network
Hu S, Offenbacher AR, Thompson EM, Gee CL, Wilcoxen J, Carr CAM, Prigozhin DM, Yang V, Alber T, Britt RD, Fraser JS, Klinman JP
JACS, 2019
- PMID: 30645119
- PMCID: PMC6353671
- Full Text
- Klinman lab @ UC Berkeley
Access the paper
Additional Link
qFit-ligand reveals widespread conformational heterogeneity of drug-like molecules in X-ray electron density maps
van Zundert GCP*, Hudson BM*, Oliveira SHP, Keedy DA, Fonseca R, Heliou A, Suresh P, Borrelli K, Day T, Fraser JS, van den Bedem H
Journal of Medicinal Chemistry, 2018
- PMID: 30457858
- PMCID: PMC6820680
- BioRxiv Preprint: 253419
- Full Text
- GitHub Repository: ExcitedStates/qfit_ligand (qFit-ligand)
- Henry van den Bedem @ Stanford University
- Schrodinger Inc.
- Paper Submission Celebration Photo
- Science Magazine: In the Pipeline-More Than One, And Maybe More Than That
- Practical Fragments: (Not) getting misled by crystal structures - part 5 – conformational heterogeneity
Access the paper
Additional Links
Model selection for biological crystallography
Babcock NS, Keedy DA, Fraser JS, Sivak DA
Biorxiv, 2018
- BioRxiv Preprint: 448795
- David Sivak @ Simon Fraser University
- Paper Submission Celebration Tweetstorm
Access the paper
Additional Links
Extending chemical perturbations of the ubiquitin fitness landscape in a classroom setting reveals new constraints on sequence tolerance
Mavor D, Barlow KA, Asarnow D, Birman Y, Britain D, Chen W, Green EM, Kenner LR, Mensa B, Morinishi LS, Nelson CA, Poss EM, Suresh P, Tian R, Arhar T, Ary BE, Bauer DP, Bergman ID, Brunetti RM, Chio CM, Dai SA, Dickinson MS, Elledge SK, Helsell CV M, Hendel NL, Kang E, Kern N, Khoroshkin MS, Kirkemo LL, Lewis GR, Lou K, Marin WM, Maxwell AM, McTigue PF, Myers-Turnbull D, Nagy TL, Natale AM, Oltion K, Pourmal S, Reder GK, Rettko NJ, Rohweder PJ, Schwarz DMC, Tan SK, Thomas PV, Tibble RW, Town JP, Tsai MK, Ugur FS, Wassarman DR, Wolff AM, Wu TS, Bogdanoff D, Li J, Thorn KS, O’Conchúir S, Swaney DL, Chow ED, Madhani HD, Redding S, Bolon DN, Kortemme T, DeRisi JL, Kampmann M, Fraser JS
Biology Open, 2018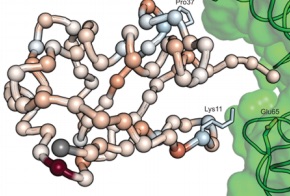
- PMID: 30037883
- PMCID: PMC6078352
- BioRxiv Preprint: 139352
- Full Text
- GitHub Repository: fraser-lab/PUBS (PUBS 2018)
- Course Syllabus: Physical Underpinnings of Biological Systems
Access the paper
Additional Link
An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering
Keedy DA*, Hill ZB*, Biel JT, Kang E, Rettenmaier TJ, Brandao-Neto J, Pearce NM, von Delft F, Wells JA, Fraser JS
eLife, 2018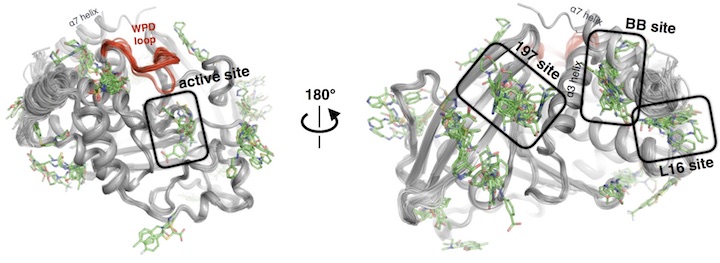
- PMID: 29877794
- PMCID: PMC6039181
- BioRxiv Preprint: 218966
- Full Text
- Deposited Structures: 6B90, 6B8E, 6B8T, 6B8X, 6B8Z, 6BAI, 6B95, 5QDE, 5QDF, 5QDG, 5QDH, 5QDI, 5QDJ, 5QDK, 5QDL, 5QDM, 5QDN, 5QDO, 5QDP, 5QDQ, 5QDR, 5QDS, 5QDT, 5QDU, 5QDV, 5QDW, 5QDX, 5QDY, 5QDZ, 5QE0, 5QE1, 5QE2, 5QE3, 5QE4, 5QE5, 5QE6, 5QE7, 5QE8, 5QE9, 5QEA, 5QEB, 5QEC, 5QED, 5QEE, 5QEF, 5QEG, 5QEH, 5QEI, 5QEJ, 5QEK, 5QEL, 5QEM, 5QEN, 5QEO, 5QEP, 5QEQ, 5QER, 5QES, 5QET, 5QEU, 5QEV, 5QEW, 5QEX, 5QEY, 5QEZ, 5QF0, 5QF1, 5QF2, 5QF3, 5QF4, 5QF5, 5QF6, 5QF7, 5QF8, 5QF9, 5QFA, 5QFB, 5QFC, 5QFD, 5QFE, 5QFF, 5QFG, 5QFH, 5QFI, 5QFJ, 5QFK, 5QFL, 5QFM, 5QFN, 5QFO, 5QFP, 5QFQ, 5QFR, 5QFS, 5QFT, 5QFU, 5QFV, 5QFW, 5QFX, 5QFY, 5QFZ, 5QG0, 5QG1, 5QG2, 5QG3, 5QG4, 5QG5, 5QG6, 5QG7, 5QG8, 5QG9, 5QGA, 5QGB, 5QGC, 5QGD, 5QGE, 5QGF
- Zenodo Record: 1044103
- XChem - Fragment Screening @ Diamond Light Source
- PANDDA code on Bitbucket
- Wells lab @ UC San Francisco
- OPIG Journal Club
- Paper Submission Celebration Photo
Access the paper
Additional Links
Bringing diffuse X-ray scattering into focus
Wall ME, Wolff AM, Fraser JS
Current Opinion in Structural Biology, 2018
- PMID: 29455056
- PMCID: PMC6078797
- BioRxiv Preprint: 219113
- Full Text
- GitHub Repository: fraser-lab/diffuse_scattering (Sematura)
- Michael Wall @ Los Alamos National Lab
- Paper Submission Celebration Photo
Access the paper
Additional Links
Rescue of conformational dynamics in enzyme catalysis by directed evolution
Otten R*, Liu L*, Kenner LR, Clarkson MW, Mavor D, Tawfik DS, Kern D, Fraser JS
Nature Communications, 2018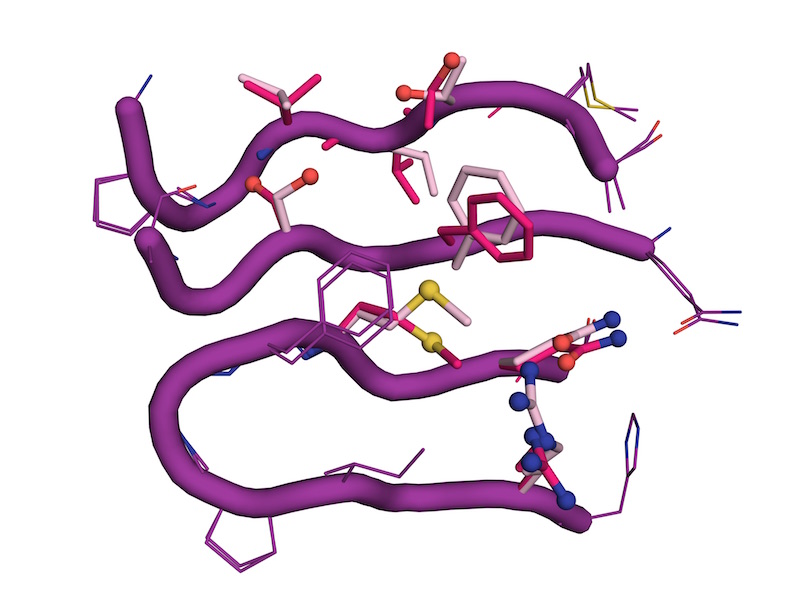
- PMID: 29615624
- PMCID: PMC5883053
- BioRxiv Preprint: 185009
- Full Text
- Deposited Structures: 5WC7, 6BTA
- Kern lab @ Brandeis University
- Faculty Opinions
Access the paper
Additional Links
XFEL structures of the influenza M2 proton channel: Room temperature water networks and insights into proton conduction
Thomaston JL, Woldeyes RA, Nakane T, Yamashita A, Tanaka T, Koiwai K, Brewster AS, Barad BA, Chen Y, Lemmin T, Uervirojnangkoorn M, Arima T, Kobayashi J, Masuda T, Suzuki M, Sugahara M, Sauter NK, Tanaka R, Nureki O, Tono K, Joti Y, Nango E, Iwata S, Yumoto F, Fraser JS, DeGrado WF
PNAS, 2017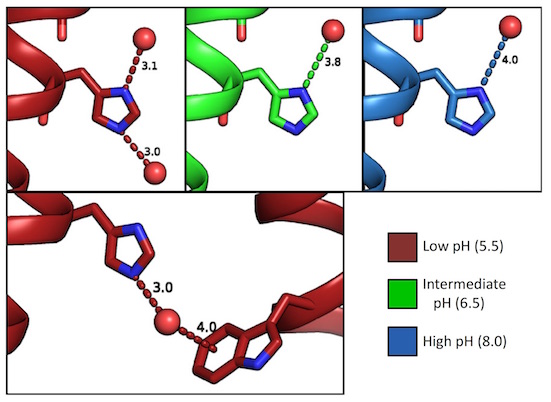
- PMID: 28835537
- PMCID: PMC5754760
- Full Text
- Deposited Structures: 5JOO, 5UM1, 5TTC
- DeGrado lab @ UC San Francisco
- SACLA XFEL
Access the paper
Additional Links
Allosteric inhibitors, crystallography and comparative analysis reveal network of coordinated movement across human herpesvirus proteases
Acker TM, Gable JE, Bohn MF, Jaishankar P, Thompson MC, Fraser JS, Renslo AR, Craik CS
JACS, 2017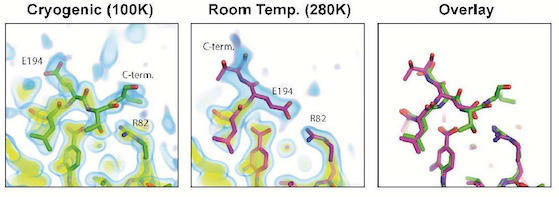
Hydrogen–Deuterium exchange of Lipoxygenase uncovers a relationship between distal, solvent exposed protein motions and the thermal activation barrier for catalytic proton-coupled electron tunneling
Offenbacher AR , Hu S, Poss EM, Carr CAM , Scouras AD, Prigozhin DM, Iavarone AT, Palla A, Alber T, Fraser JS, Klinman JP
ACS Central Science, 2017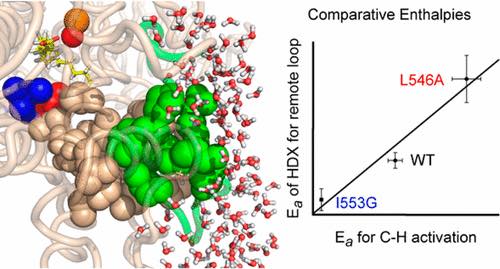
- PMID: 28691068
- PMCID: PMC5492416
- Full Text
- Deposited Structures: 5TQO, 5TQP
- Klinman lab @ UC Berkeley
Access the paper
Additional Link
Flexibility and design: conformational heterogeneity along the evolutionary trajectory of a redesigned ubiquitin
Biel JT, Thompson MC, Cunningham CN, Corn JE, Fraser JS
Structure, 2017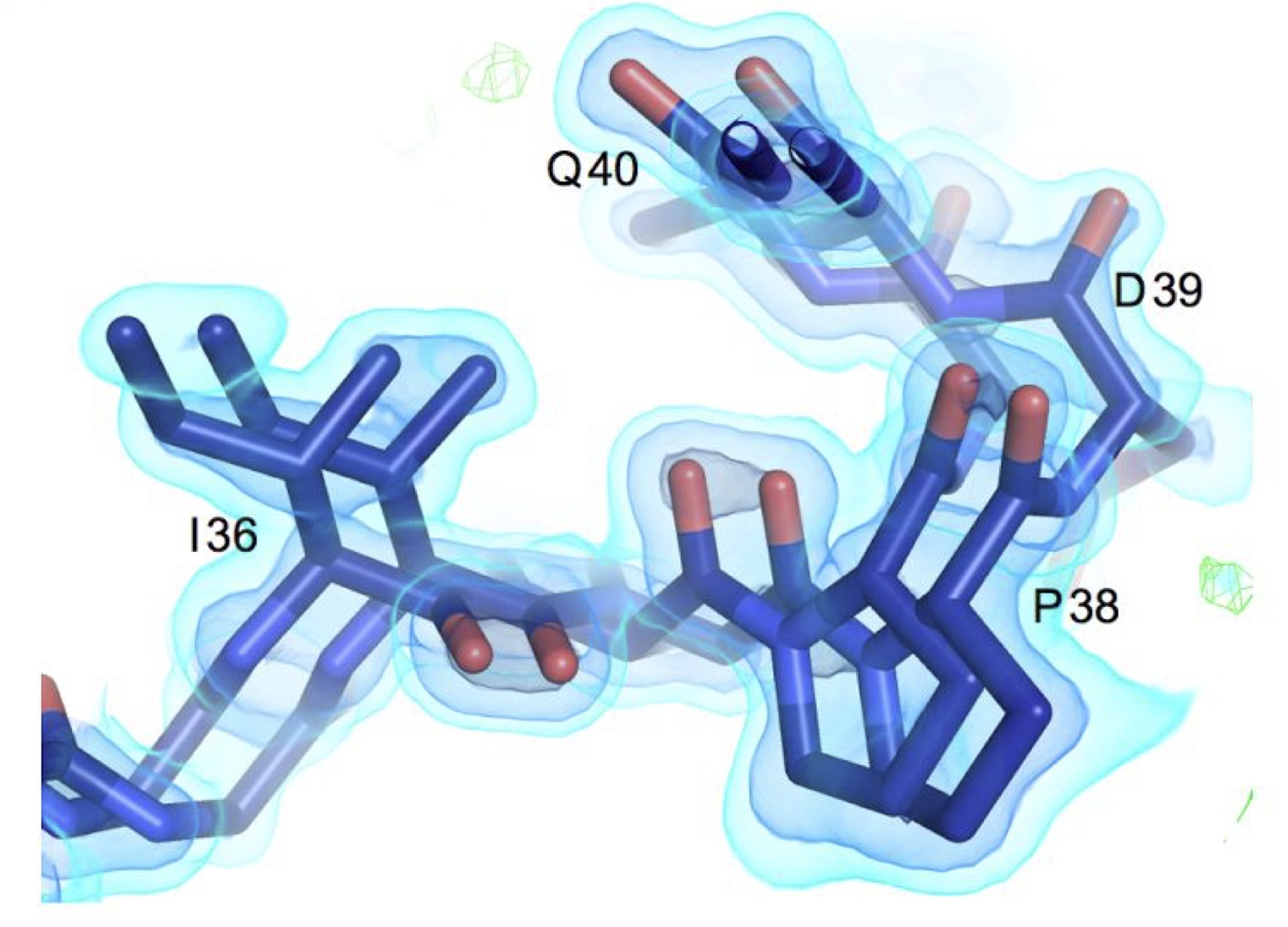
- PMID: 28416112
- PMCID: PMC5415430
- BioRxiv Preprint: 081646
- Full Text
- Online Datasets:
- Deposited Structures: 5TOF, 5TOG
- Genentech
- Paper Submission Celebration Photo
- Cell: Room Temperature X-Ray Crystallography Reveals Conformational Heterogeneity of Engineered Proteins
Access the paper
Additional Links
Cytidine deaminase efficiency of the lentiviral viral restriction factor APOBEC3C correlates with dimerization
Adolph MB, Ara A, Feng Y, Wittkopp CJ, Emerman M, Fraser JS, Chelico L
Nucleic Acids Research, 2017
- PMID: 28158858
- PMCID: PMC5389708
- Full Text
- Linda Chelico @ University of Saskatchewan
- HARC Center @ UC San Francisco
Access the paper
Additional Links
Conformational variation of proteins at room temperature is not dominated by radiation damage
Russi S, González A, Kenner LR, Keedy DA, Fraser JS, van den Bedem H
Journal of Synchrotron Radiation, 2017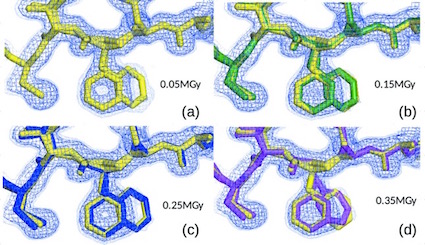
- PMID: 28009548
- PMCID: PMC5182021
- Full Text
- Henry van den Bedem @ Stanford University
Access the paper
Additional Link
Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta
Wang RYR, Song Y, Barad BA, Cheng Y, Fraser JS, DiMaio F
eLife, 2016
- PMID: 27669148
- PMCID: PMC5115868
- BioRxiv Preprint: 050286
- Full Text
- DiMaio lab @ University of Washington
Access the paper
Additional Link
Preprints for the life sciences
Berg JM, Bhalla N, Bourne PE, Chalfie M, Drubin DG, Fraser JS, Greider CW, Hendricks M, Jones C, Kiley R, King S, Kirschner MW, Krumholz HM, Lehmann R, Leptin M, Pulverer B, Rosenzweig B, Spiro JE, Stebbins M, Strasser C, Swaminathan S, Turner P, Vale RD, VijayRaghavan K, Wolberger C
Science, 2016
- PMID: 27199406
- ASAPbio 2016 meeting
Access the paper
Additional Link
Determination of Ubiquitin fitness landscapes under different chemical stresses in a classroom setting
Mavor D, Barlow KA, Thompson S, Barad BA, Bonny AR, Cario CL, Gaskins G, Liu Z, Deming L, Axen SD, Caceres E, Chen W, Cuesta A, Gate R, Green EM, Hulce KR, Ji W, Kenner LR, Mensa B, Morinishi LS, Moss SM, Mravic M, Muir RK, Niekamp S, Nnadi CI, Palovcak E, Poss EM, Ross TD, Salcedo E, See S, Subramaniam M, Wong AW, Li J, Thorn KS, Conchúir SÓ, Roscoe BP, Chow ED, DeRisi JL, Kortemme T, Bolon DN, Fraser JS
eLife, 2016
- PMID: 27111525
- PMCID: PMC4862753
- BioRxiv Preprint: 025452
- Full Text
- GitHub Repository: fraser-lab/PUBS2014
- Course Syllabus: Physical Underpinnings of Biological Systems
- Paper Submission Celebration Photo
- eLIFEdigest: Yeast in a class of their own
Access the paper
Additional Links
Measuring and modeling diffuse scattering in protein X-ray crystallography
Van Benschoten AH, Liu L, Gonzalez A, Brewster AS, Sauter NK, Fraser JS, Wall ME
PNAS, 2016
- PMID: 27035972
- PMCID: PMC4839442
- BioRxiv Preprint: 033746
- Full Text
- Online Datasets:
- Deposited Structures: 5F6M, 5F66
- Michael Wall @ Los Alamos National Lab
- Paper Submission Celebration Photo
Access the paper
Additional Links
Data publication with the structural biology data grid supports live analysis
Meyer PA, Socias S, Key J, Ransey E, Tjon EC, Buschiazzo A, Lei M, Botka C, Withrow J, Neau D, Rajashankar K, Anderson KS, Baxter RH, Blacklow SC, Boggon TJ, Bonvin AMJJ, Borek D, Brett TJ, Caflisch A, Chang C, Chazin WJ, Corbett KD, Cosgrove MS, Crosson S, Dhe-Paganon S, Cera ED, Drennan CL, Eck MJ, Eichman BF, Fan QR, Ferré-D’Amaré AR, Fromme JC, Garcia KC, Gaudet R, Gong P, Harrison SC, Heldwein EE, Jia Z, Keenan RJ, Kruse AC, Kvansakul M, McLellan JS, Modis Y, Nam Y, Otwinowski Z, Pai EF, Pereira PJB, Petosa C, Raman CS, Rapoport TA, Roll-Mecak A, Rosen MK, Rudenko G, Schlessinger J, Schwartz TU, Shamoo Y, Sondermann H, Tao YJ, Tolia NH, Tsodikov OV, Westover KD, Wu H, Foster I, Fraser JS, Maia FRNC, Gonen T, Kirchhausen T, Diederichs K, Crosas M, Sliz P
Nature Communications, 2016
- PMID: 26947396
- PMCID: PMC4786681
- Full Text
- Online Dataset: doi: 10.15785/SBGRID/68
- Deposited Structure: 4YUO
- SBGrid
Access the paper
Additional Link
CryptoSite: Expanding the druggable proteome by characterization and prediction of cryptic binding sites
Cimermancic P, Weinkam P, Rettenmaier TJ, Bichmann L, Keedy DA, Woldeyes RA, Schneidmann D, Demerdash ONA, Mitchell JC, Wells JA, Fraser JS, Sali A
Journal of Molecular Biology, 2016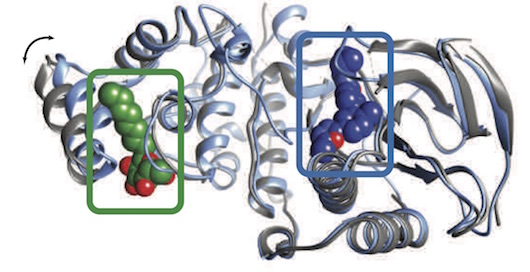
- PMID: 26854760
- PMCID: PMC4794384
- Full Text
- CryptoSite
- Paper Submission Celebration Photo
- F1000 Evaluation
- Science Magazine: More Binding Sites Than Are Dreamt of in Your Philosophy
Access the paper
Additional Links
High-density grids for efficient data collection from multiple crystals
Baxter EL, Aguila L, Alonso-Mori R, Barnes CO, BonaguraCA, Brehmer W, Brunger AT, Calero G, Caradoc-Davies TT, Chatterjee R, Degrado WF, Fraser JS, Ibrahim M, Kern J, Kobilka BK, Kruse AC, Larsson KM, Lemke HT, Lyubimov AY, Manglik A, McPhillips SE, Norgren E, Pang SS, Soltis SM, Song J, Thomaston J, Tsai Y, Weis WI, Woldeyes RA, Yachandra V, Yano J, Zouni A, Cohen AE
Acta Crystallographica D, 2016
- PMID: 26894529
- PMCID: PMC4756618
- Full Text
- LCLS XPP Beamline
Access the paper
Additional Link
High resolution structures of the M2 proton channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction
Thomaston JL, Alfonso-Prieto M, Woldeyes RA, Fraser JS, Klein ML, Fiorin G, DeGrado WF
PNAS, 2015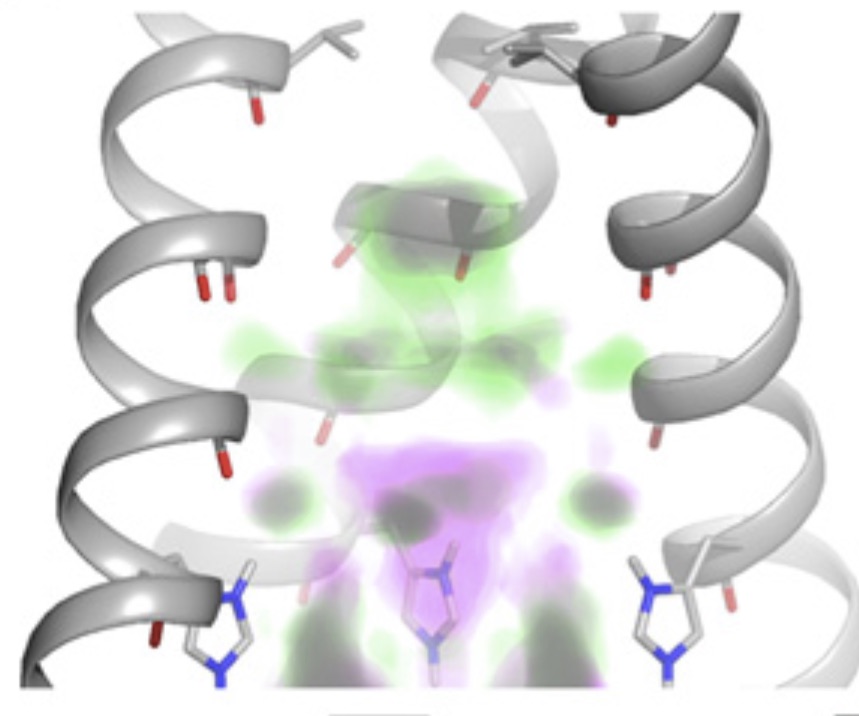
- PMID: 26578770
- PMCID: PMC4655559
- Full Text
- Deposited Structures: 4QK7, 4QKC, 4QKL, 4QKM
- DeGrado lab @ UC San Francisco
- Paper Submission Celebration Photo
Access the paper
Additional Links
Exposing hidden alternative backbone conformations in X-ray crystallography using qFit
Keedy DA, Fraser JS, van den Bedem H
PLOS Computational Biology, 2015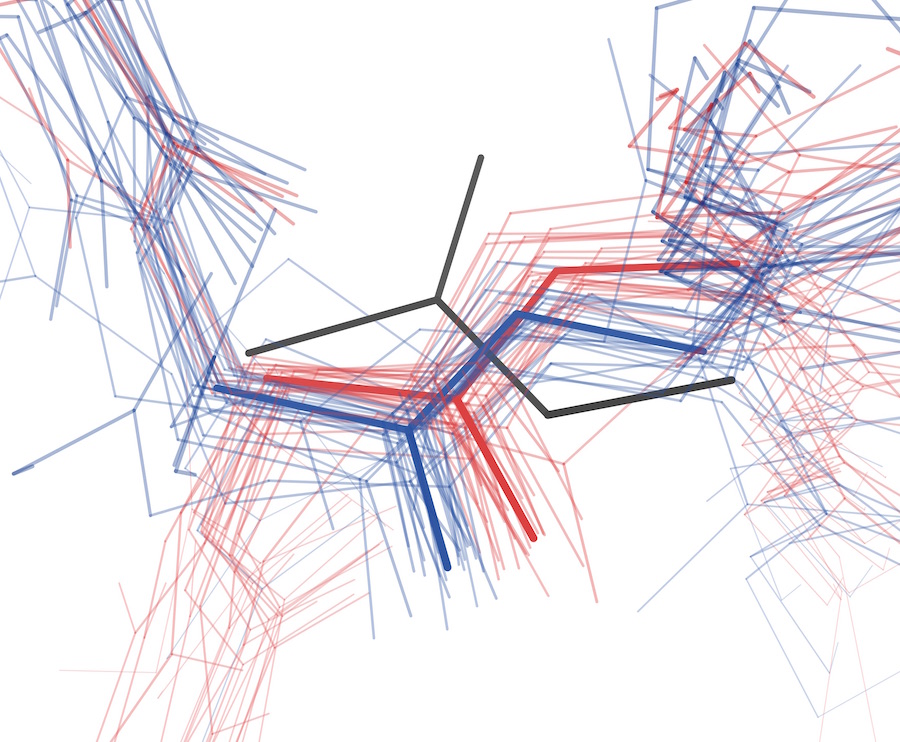
- PMID: 26506617
- PMCID: PMC4624436
- BioRxiv Preprint: 018879
- Full Text
- Zenodo Record: 7182718 (Coordinates for peptide flip clusters)
- qFit code
- Henry van den Bedem @ Stanford University
- qFit Server @ SLAC
- Paper Submission Celebration Photo
Access the paper
Additional Links
Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography
Keedy DA*, Kenner LR*, Warkentin M*, Woldeyes RA*, Hopkins JB, Thompson MC, Brewster AS, Van Benschoten AH, Baxter EL, Uervirojnangkoorn M, McPhillips SE, Song J, Alonso-Mori R, Holton JM, Weis WI, Brunger AT, Soltis SM, Lemke H, Gonzalez A, Sauter NK, Cohen AE, van den Bedem H, Thorne RE, Fraser JS
eLife, 2015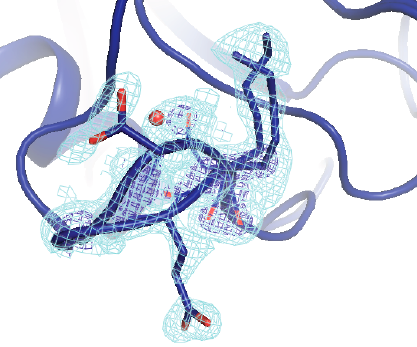
- PMID: 26422513
- PMCID: PMC4721965
- BioRxiv Preprint: 016733
- Full Text
- Online Dataset: doi: 10.15785/SBGRID/68
- Deposited Structures: 4YUG, 4YUH, 4YUI, 4YUJ, 4YUK, 4YUL, 4YUM, 4YUN, 4YUO, 4YUP
- Thorne lab @ Cornell University
- Henry van den Bedem @ Stanford University
- LCLS XPP Beamline
- Paper Submission Celebration Photo
Access the paper
Additional Links
EMRinger: Side-chain-directed model and map validation for 3D electron cryomicroscopy
Barad BA, Echols N, Wang RYR, Cheng Y, DiMaio F, Adams PD, Fraser JS
Nature Methods, 2015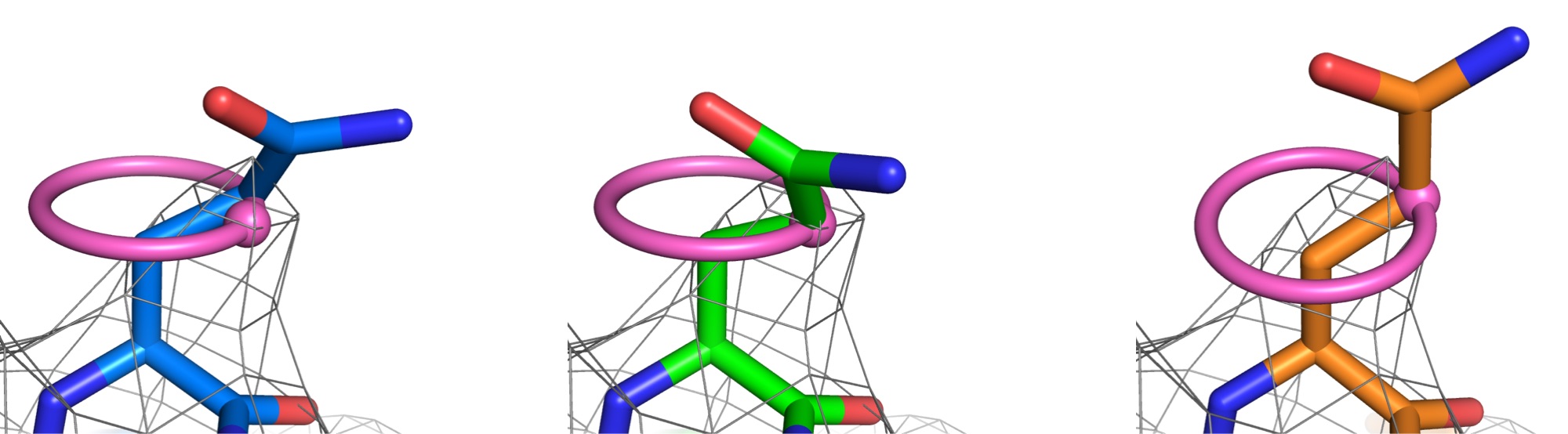
- PMID: 26280328
- PMCID: PMC4589481
- BioRxiv Preprint: 014738
- Full Text
- Deposited Structures: 3J9I, 3J9J
- GitHub Repository: fraser-lab/EMRinger
- Paper Submission Celebration Photo
Access the paper
Additional Link
Predicting X-ray diffuse scattering from translation libration screw structural ensembles
Van Benschoten AH, Afonine PV, Terwilliger TC, Wall ME, Jackson CJ, Sauter NK, Adams PD, Urzhumtsev A, Fraser JS
Acta Crystallographica D, 2015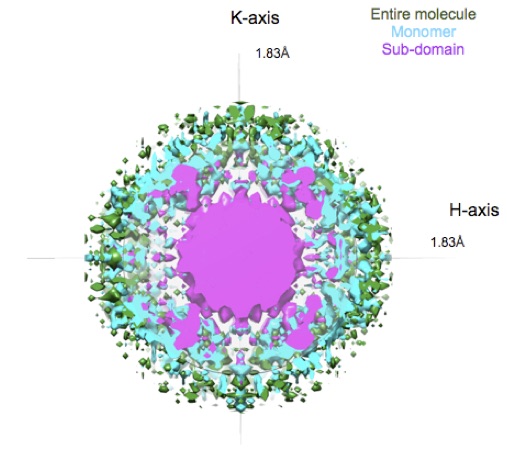
- PMID: 26249347
- PMCID: PMC4528799
- BioRxiv Preprint: 012955
- Full Text
- Paper Submission Celebration Photo
- Summary by iUCr
Access the paper
Additional Links
From deep TLS validation to ensembles of atomic models built from elemental motions
Urzhumtsev A, Afonine PV, Van Benschoten AH, Fraser JS, Adams PD
Acta Crystallographica D, 2015
- PMID: 26249348
- PMCID: PMC4528800
- BioRxiv Preprint: 012930
- Full Text
- Paper Submission Celebration Photo
- Addenda and corrigendum
Access the paper
Additional Links
One crystal, two temperatures – cryocooling penalties alter ligand binding to transient protein sites
Fischer M, Shoichet BK, Fraser JS
ChemBioChem, 2015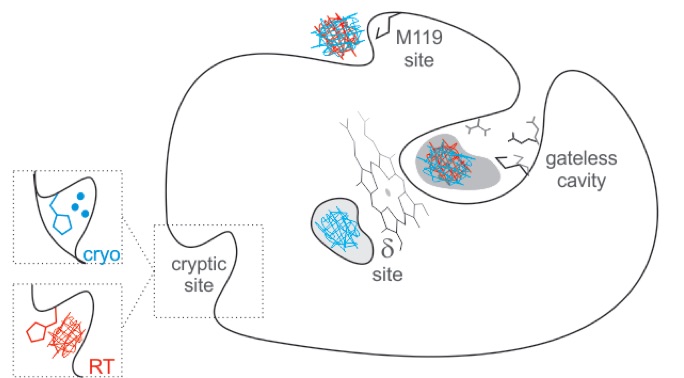
- PMID: 26032594
- PMCID: PMC4539595
- Full Text
- Deposited Structures: 4XV7, 4XV6, 4XV5, 4XV4, 4XVA, 4XVB
- Shoichet lab @ UC San Francisco
- Paper Submission Celebration Photo
- F1000 Evaluation
- Cover Photo: A Song of Fire and Ice
Access the paper
Additional Links
Negative epistasis and evolvability in TEM-1 β-lactamase – The thin line between an enzyme’s conformational freedom and disorder
Dellus-Gur E, Elias M, Caselli E, Prati F, Salverda ML, de Visser JA, Fraser JS, Tawfik DS
Journal of Molecular Biology, 2015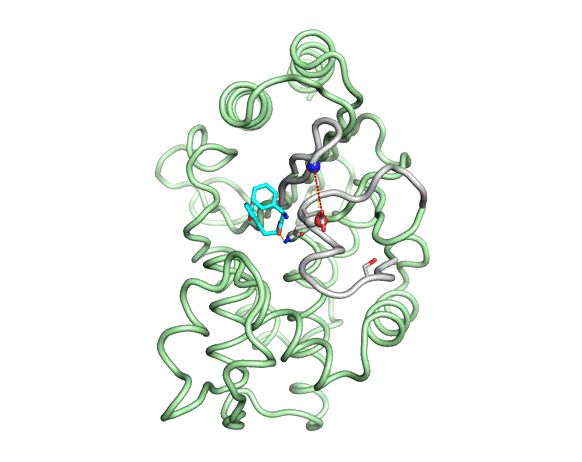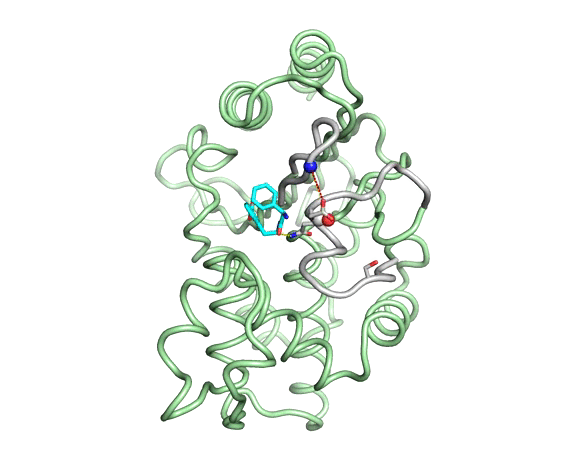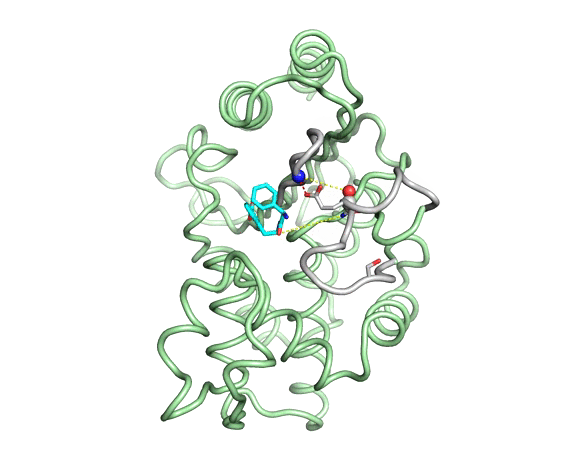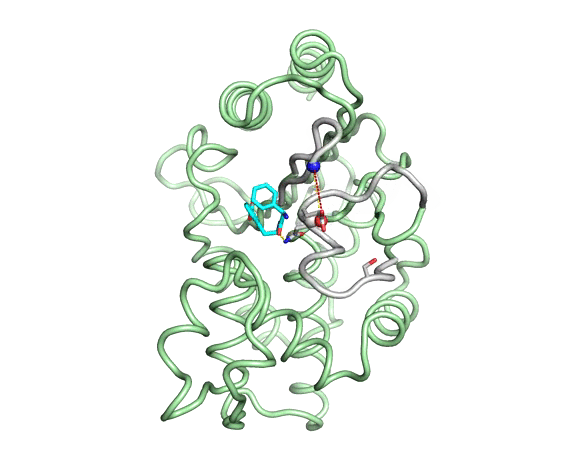
Lineage-specific viral hijacking of non-canonical E3 Ubiquitin Ligase cofactors in the evolution of Vif anti-APOBEC3 activity
Kane JR, Stanley DJ, Hultquist JF, Johnson JR, Mietrach N, Binning JM, Jonsson SR, Barelier S, Newton BW, Johnson TL, Franks-Skiba KE, Li M, Brown WL, Gunnarsson HI, Adalbjornsdottir A, Fraser JS, Harris RS, Andresdottir V, Gross JD, Krogan NJ
Cell Reports, 2015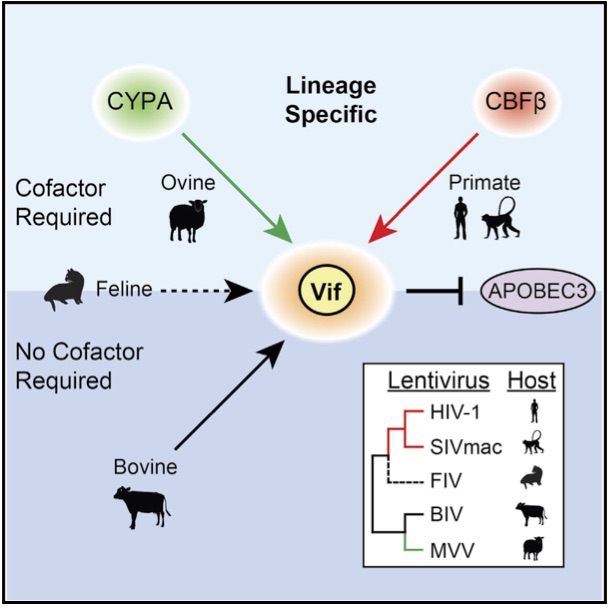
- PMID: 25981045
- PMCID: PMC4613747
- Full Text
- Gross lab @ UC San Francisco
- Krogan lab @ UC San Francisco
- HARC Center @ UC San Francisco
Access the paper
Additional Links
Integrative, dynamic structural biology at atomic resolution — it’s about time
van den Bedem H, Fraser JS
Nature Methods, 2015
- PMID: 25825836
- PMCID: PMC4457290
- Full Text
- Henry van den Bedem @ Stanford University
Access the paper
Additional Link
Keep on moving: discovering and perturbing the conformational dynamics of enzymes
Bhabha G, Biel JT, Fraser JS
Accounts of Chemical Research, 2015
- PMID: 25539415
- PMCID: PMC4334266
- Full Text
- Paper Submission Celebration Photo
Access the paper
Additional Link
Conformational dynamics of a crystalline protein from microsecond-scale molecular dynamics simulations and diffuse X-Ray scattering
Wall ME, VanBenschoten AH, Sauter NK, Adams PD, Fraser JS, Terwilliger TC
PNAS, 2014
- PMID: 25453071
- PMCID: PMC4273327
- Full Text
- Deposited Structure: 4WOR
- Michael Wall @ Los Alamos National Lab
- Paper Submission Celebration Photo
Access the paper
Additional Links
Discovery and characterization of two tryptamine-producing decarboxylases from the gut microbiota
Williams BB, VanBenschoten AH, Cimermancic P, Donia MS, Zimmermann M, Taketani M, Ishihara A, Kashyap PC, Fraser JS, Fischbach MA
Cell Host and Microbe, 2014
- PMID: 25263219
- PMCID: PMC4260654
- Full Text
- Deposited Structures: 4OBU, 4OBV
- Fischbach lab @ Stanford University
- Paper Submission Celebration Photo
- Cell Host & Microbe Podcast
- Science Signaling: Bacteria May Mess with Your Mind
- Nature Reviews Microbiology: Bacteria and the brain
Access the paper
Additional Links
E pluribus unum, no more: from one crystal, many conformations
Woldeyes RA, Sivak DA, Fraser JS
Current Opinion in Structural Biology, 2014
- PMID: 25113271
- PMCID: PMC4253534
- Full Text
- David Sivak @ Simon Fraser University
- Paper Submission Celebration Photo
Access the paper
Additional Links
Crystal cryocooling distorts conformational heterogeneity in a model Michaelis complex of DHFR
Keedy DA, van den Bedem H, Sivak DA, Petsko GA, Ringe D, Wilson MA, Fraser JS
Structure, 2014
- PMID: 24882744
- PMCID: PMC4082491
- Full Text
- Deposited Structures: 4PSS, 4PST, 4PSY, 4PSZ, 4PTJ, 4P3Q, 4P3R, 4PTH
- Wilson lab @ University of Nebraska-Lincoln
- Petsko/Ringe lab @ Brandeis University
- Paper Submission Celebration Photo
- F1000 Evaluation
Access the paper
Additional Links
Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery
Fischer M*, Coleman RG*, Fraser JS, Shoichet BK
Nature Chemistry, 2014
- PMID: 24950326
- PMCID: PMC4144196
- Full Text
- Deposited Structures: 4OQ7, 4NVA, 4NVB, 4NVC, 4NVD, 4NVE, 4NVF, 4NVG, 4NVH, 4NVI, 4NVJ, 4NVK, 4NVL, 4NVM, 4NVN, 4NVO
- DOCK Software
- Shoichet lab @ UC San Francisco
- News and Views - Ligand discovery, Docking points
- Nature Chemistry Cover
- Cover Explanation
Access the paper
Additional Links
Diffuse X-ray scattering to model protein motions
Wall ME, Adams PD, Fraser JS, Sauter NK
Structure, 2014
- PMID: 24507780
- PMCID: PMC4070675
- Full Text
- Meeting Slides and Website
Access the paper
Additional Link
Integrated description of protein dynamics from room-temperature X-ray crystallography and NMR
Fenwick RB, van den Bedem H, Fraser JS, Wright PE
PNAS, 2014
- PMID: 24474795
- PMCID: PMC3910589
- Full Text
- Deposited Structures: 4NX6, 4NX7
- Python Scripts
- Wright lab @ Scripps Research
- F1000 Evaluation
Access the paper
Additional Links
Protein structural ensembles are revealed by redefining X-ray electron density noise
Lang PT, Holton JM, Fraser JS, Alber T
PNAS, 2014
- PMID: 24363322
- PMCID: PMC3890839
- Full Text
- Deposited Structure: 4NX4
- END/RAPID Website
Access the paper
Additional Link
From structure to systems: high-resolution, quantitative genetic analysis of RNA polymerase II
Braberg H, Jin H, Moehle E, Chan YA, Wang S, Shales M, Benschop JJ, Morris JH, Fraser JS, Qiu C, Hu F, Tang LK, Holstege FCP, Hieter P, Guthrie C, Kaplan CD, Krogan NJ
Cell, 2013
- PMID: 23932120
- PMCID: PMC3932829
- Full Text
- Krogan lab @ UC San Francisco
- F1000 Evaluation
- UCSF: Innovative Method to Profile and Predict the Behavior of Proteins
- Nature: Pol II Interactions in Detail
Access the paper
Additional Links
Automated identification of functional dynamic contact networks from X-ray crystallography
van den Bedem H, Bhabha G, Yang K, Wright PE, Fraser JS
Nature Methods, 2013
- PMID: 23913260
- PMCID: PMC3760795
- Full Text
- Deposited Structures: 4KJJ, 4KJK, 4KJL
- CONTACT Website
- Henry van den Bedem @ Stanford University
- Wright lab @ Scripps Research
- F1000 Evaluation
- News and Views - Vizualizing Networks of Protein Mobility
- Nature Structural and Molecular Biology: Crystal Clear Dynamics
- Nature Chemical Biology: Shake it up, baby
- SBKB Drug Discovery: Identifying Dynamic Networks by CONTACT
- SLAC: How Proteins Shift into Working Mode
- Youtube: CONTACT
Access the paper
Additional Links
Flexible backbone sampling methods to model and design protein alternative conformations
Ollikainen N, Smith CA, Fraser JS, Kortemme T
Methods in Enzymology, 2013
- PMID: 23422426
- PMCID: PMC3750959
- Full Text
- Backrub Server @ Kortemme lab
- Supplementary Material
- Kortemme lab @ UC San Francisco
Access the paper
Additional Links
From systems to structure: bridging networks and mechanism
Fraser JS, Gross JD, Krogan NJ
Nature Methods, 2013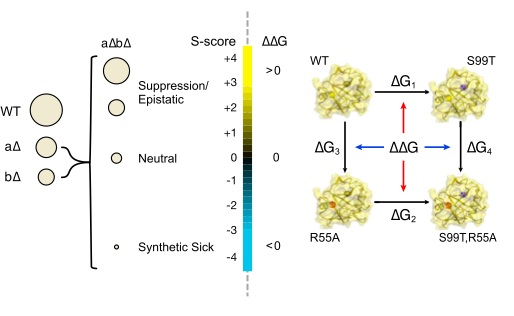
- PMID: 23352243
- PMCID: PMC3558917
- Full Text
- Gross lab @ UC San Francisco
- Krogan lab @ UC San Francisco
- Molecular Cell: Celebrating 15 years of New Connections
Access the paper
Additional Links
CheShift-2 resolves a local inconsistency between two X-ray crystal structures
Vila JA, Sue SC, Fraser JS, Scheraga HA, Dyson HJ
Journal of Biomolecular NMR, 2012
- PMID: 22945426
- PMCID: PMC3471536
- Full Text
- CheShift-2 Server @ Universidad Nacional de San Luis
- Dyson lab @ Scripps Research
- Scheraga lab @ Cornell University
Access the paper
Additional Links
Systematic functional prioritization of protein posttranslational modifications
Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J, Lim WA, Fraser JS, Frydman J, Krogan NJ
Cell, 2012
- PMID: 22817900
- PMCID: PMC3404735
- Full Text
- PTMfunc database
- Krogan lab @ UC San Francisco
- Nature Methods: Making PTMs a priority
- Pedro's Public Ramblings: Evolution and Function of Post-translational Modifications
Access the paper
Additional Links
Control of protein signaling using a computationally designed GTPase/GEF orthogonal pair
Kapp GT*, Liu S*, Stein A, Wong DT, Reményi A, Yeh BJ, Fraser JS, Taunton J, Lim WA, Kortemme T
PNAS, 2012
- PMID: 22403064
- PMCID: PMC3325720
- Full Text
- Deposited Structure: 3QBV
- Kortemme lab @ UC San Francisco
- PNAS: Designing orthogonal signaling pathways: how to fit in with the surroundings.
Access the paper
Additional Links
Accessing protein conformational ensembles by room-temperature X-ray crystallography
Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T
PNAS, 2011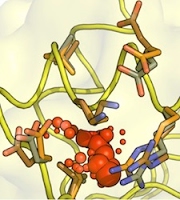
- PMID: 21918110
- PMCID: PMC3182744
- Full Text
- Deposited Structure: 3TGP
- qFIT server @ SLAC
- F1000 evaluation
- SBKB: Not so cool
- PNAS: In this issue
- SLAC: Proteins better analyzed at room temperature
- PLoS - Computational Biology: Postcard based on a talk just prior to publication
Access the paper
Additional Links
Mining electron density for functionally relevant protein polysterism in crystal structures
Fraser JS, Jackson CJ
Cellular and Molecular Life Sciences, 2010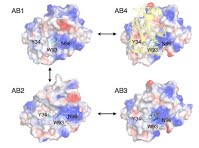
- PMID: 21190057
- PMCID: PMC3092063
- Full Text
- Jackson lab @ Australian National University
Access the paper
Additional Link
The tumor-associated EpCAM regulates morphogenetic movements through intracellular signaling
Maghzal N*, Vogt E*, Reintsch W, Fraser JS, Fagotto F
Journal of Cell Biology, 2010
- PMID: 20974811
- PMCID: PMC3003323
- Full Text
- Fagotto lab @ McGill University
Access the paper
Additional Link
Automated electron-density sampling reveals widespread conformational polymorphism in proteins
Lang PT*, Ng HL*, Fraser JS, Corn JE, Echols N, Sales M, Holton JM, Alber T
Protein Science, 2010
- PMID: 20499387
- PMCID: PMC2974833
- Full Text
- Ringer Website @ Lawrence Berkeley National Lab
- Ringer is now also available as an alpha feature in the PHENIX GUI or on the command line as mmtbx.ringer
- F1000 evaluation
- Protein Science: Peripatetic proteins
- QB3: QB3 structural biologists make algorithm available on the web
Access the paper
Additional Links
Hidden alternative structures of proline isomerase essential for catalysis
Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T
Nature, 2009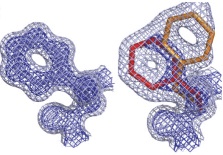
- PMID: 19956261
- PMCID: PMC2805857
- Full Text
- Deposited Structures: 3K0M, 3K0N, 3K0O, 3K0P, 3K0Q, 3K0R
- F1000 evaluation
- Nature: Editor's Summary
- QB3: QB3 scientists pin down shape-changing enzyme
- Chemical & Engineering News: Crystallographic Noise Characterizes Enzyme
- Conformational Flux: Alternate Structures and Catalysis in Cyclophilin
Access the paper
Additional Links
Immunoglobulin-Like domains on bacteriophage: weapons of modest damage?
Fraser JS, Maxwell KL, Davidson AR
Current Opinion in Microbiology, 2007
- PMID: 17765600
- Full Text
- Davidson lab @ University of Toronto
Access the paper
Additional Link
An atypical receiver domain controls the dynamic polar localization of the Myxococcus xanthus social motility protein FrzS
Fraser JS*, Merlie Jr. JP*, Echols N*, Weisfield SR, Mignot T, Wemmer DE, Zusman D, Alber T
Molecular Microbiology, 2007
- PMID: 17573816
- PMCID: PMC1974792
- Full Text
- Deposited Structures: 2GKG, 2I6F, 2NT4, 2NT3
- Zusman lab @ UC Berkeley
Access the paper
Additional Link
Ig-like domains on bacteriophages: a tale of promiscuity and deceit
Fraser JS, Yu Z, Maxwell KL, Davidson AR
Journal of Molecular Biology, 2006
- PMID: 16631788
- Full Text
- Davidson lab @ University of Toronto